Rieger’s Anomaly without Posterior Embryotoxon: A Rare Presentation
Shrinkhal1, Mood Mahesh2, Ajai Agrawal3, Ramanuj Samanta4, Anupam Singh5
1 Senior Resident, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India.
2 Junior Resident, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India.
3 Additional Professor, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India.
4 Assistant Professor, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India.
5 Additional Professor, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh, Uttarakhand, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Ajai Agrawal, Department of Ophthalmology, All India Institute of Medical Sciences, Rishikesh-249203, Uttarakhand, India.
E-mail: ajaiagrawal@rediffmail.com
Rieger’s anomaly is a rare congenital ocular defect with autosomal dominant inheritance, characterised by dysgenesis of the anterior segment. Ocular features of typical Reiger’s anomaly include a prominent anteriorly displaced Schwalbe’s line (posterior embryotoxon), iris stromal hypoplasia, corectopia, and glaucoma. An atypical presentation of Rieger’s anomaly is described in the current case report. A 26-year-old female presented with diminution of vision in right eye and mild photophobia in both eyes, since childhood. On examination, it was recognised as an atypical isolated case of Rieger’s anomaly with several classical features including segmental full thickness iris defect and ectropion uveae. This was associated with total cataract in right eye and persistent pupillary membrane in left eye, without posterior embryotoxon and glaucoma. There were no other associated ocular or systemic anomalies. Patient was operated for cataract surgery in right eye under guarded visual prognosis. The postoperative visual acuity was 3/60, signifying pre-existing amblyopia. The patient was kept on close follow-up for the development of glaucoma. This is a rare case of Anterior Segment Dysgenesis (ASD) manifesting as atypical Rieger’s anomaly without posterior embryotoxon and any systemic association, signifying the fact that posterior embryotoxon is not an essential diagnostic criterion.
Axenfeld-Rieger syndrome, Ectropion uveae, Iris aplasia, Persistent pupillary membrane
Case Report
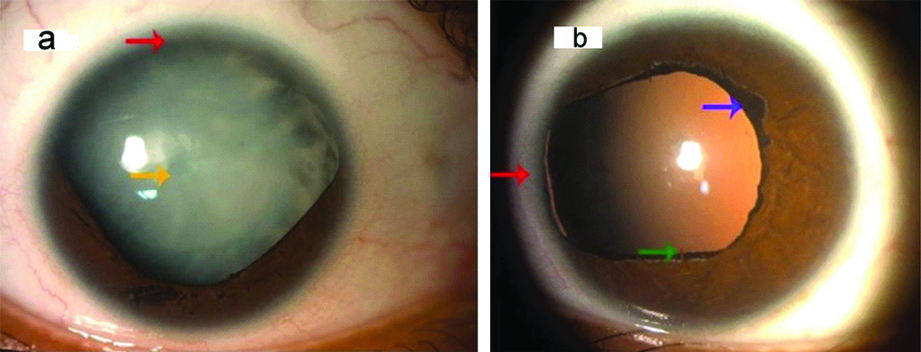
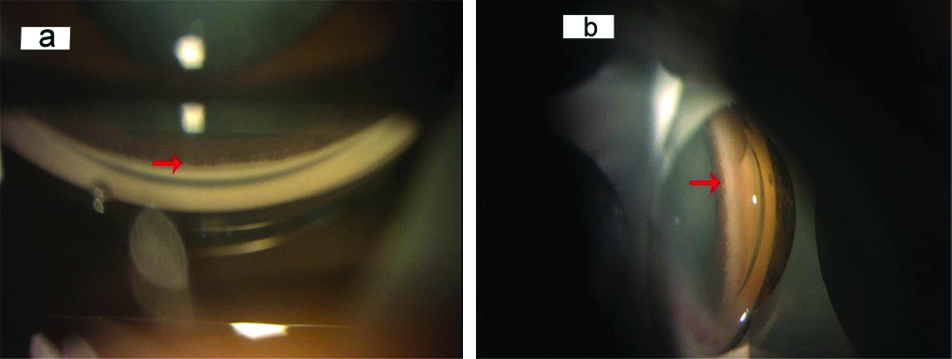
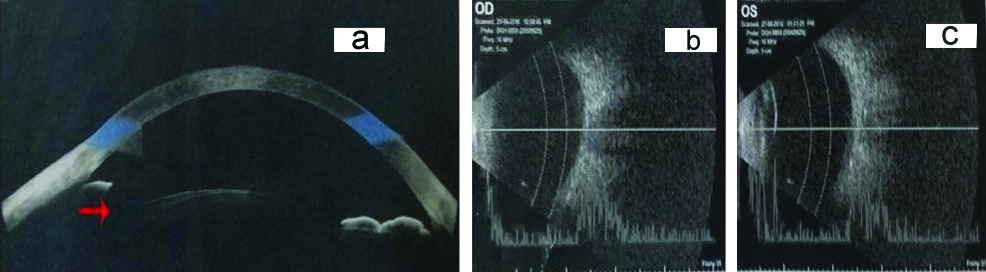
A 26-year-old female presented with chief complaints of gradual progressive painless diminution of vision in right eye and mild photophobia in both eyes since childhood and white opacity in right eye since the past 20 days. There was no history of trauma to eyes, prolonged drug intake, radiation exposure or any fever. Birth history was uneventful. General physical examination and systemic examination were normal. The visual acuity at presentation was Finger Count Close to Face (FCCF) with Projection of Rays (PR) accurate in all four quadrants in right eye and 6/12 in left eye, improving to 6/9 on pin hole on Snellen’s chart. On retinoscopy, there was absence of red reflex in right eye and in left eye, there was a refractive error of +1.50 D cylinder at 120 degrees with Best Corrected Visual Acuity (BCVA) of 6/9. The Intraocular Pressure (IOP) by Goldman applanation tonometry was 12 mmHg and 14 mmHg in right and left eye, respectively. On anterior segment examination by slit lamp, corneal diameters of right eye were 10.5 mm and 11.0 mm in horizontal and vertical axes respectively and in left eye, they were 11.0 mm and 10.0 mm in horizontal and vertical axes respectively, signifying normal corneal size. Peripheral anterior chamber depth was Van Herick grade 4 in both the eyes and there was partial aplasia of iris from 3-9 clock hours in right eye ([Table/Fig-1a]; red arrow) and from 8-10 clock hours in left eye ([Table/Fig-1b]; red arrow). Pupil had corectopia, dyscoria with very sluggish reaction to light in both eyes with presence of ectropion uveae in left eye ([Table/Fig-1b]; purple arrow) and persistent pupillary membrane in left eye ([Table/Fig-1b]; green arrow). Fundus was not visible in right eye, due to dense cataract and it was normal in left eye. Right eye also revealed total cataract ([Table/Fig-1a]; yellow arrow) with iris pigment deposits on anterior capsule of lens and left eye had Lens Opacity Classification System (LOCS) grade 1 Posterior Subcapsular Cataract (PSC). Gonioscopy revealed open angles (Shaffer’s grade 3-4) in both eyes with presence of only iris root stump in superior quadrant in right eye and nasal quadrant in left eye. There were no iridocorneal adhesions ([Table/Fig-2a,b]; red arrows). Anterior Segment Optical Coherence Tomography (AS-OCT) showed segmental absence of iris in right eye ([Table/Fig-3a]; red arrow). Axial length was 22.49 mm and 22.14 mm in right and left eye, respectively. Brightness scan (B-scan) ultrasonography revealed normal posterior segment in both the eyes [Table/Fig-3b,c]. On specular microscopy, corneal endothelial cell density was 3098 cells/mm2 with 65% hexagonal cells and a Central Corneal Thickness (CCT) of 582 microns in right eye. Corneal endothelial cell density was 2996 cells/mm2 with 73% hexagonal cells and CCT of 578 microns in left eye, suggestive of normal corneal endothelium, bilaterally.
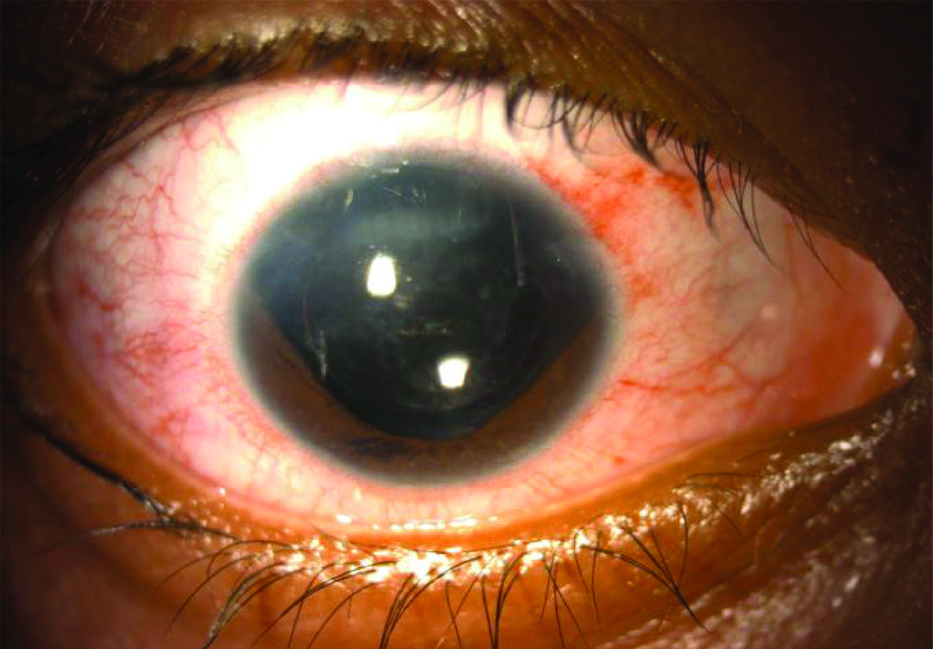
Patient underwent dental, cardiovascular and abdominal examination, which was unremarkable. A diagnosis of Atypical Rieger’s anomaly without posterior embryotoxon was made. Written, informed consent was taken from the patient for cataract surgery, as well as for sharing her clinical photographs for scientific purposes. Patient was operated for cataract in right eye, under guarded visual prognosis. Postoperatively [Table/Fig-4], the vision was 3/60 in right eye with no acceptance of glasses, signifying that right eye had amblyopia. The patient is currently under follow-up, since the past six months and has not manifested any features of glaucoma.
a) Slit lamp photograph showing partial aplasia of iris from 3-9 clock hours in right eye (red arrow) with total cataract (yellow arrow). b) Slit lamp photograph showing partial aplasia of iris from 8-10 clock hours in left eye (red arrow) with ectropion uveae (purple arrow), persistent pupillary membrane (green arrow) and LOCS grade 1 PSC.
a,b: Presence of open angle and iris root stump at sites of iris aplasia (red arrows) in gonioscopy images of right and left eye respectively.
a: Anterior segment Optical Coherence Tomography image showing segmental iris aplasia (red arrow). b,c: B-scan Ultrasonography images showing normal posterior segment in right and left eye, respectively.
Postoperative slit lamp photograph of right eye with well-centered posterior chamber intraocular lens.
Discussion
The ASD involves maldevelopment of any ocular structure in front of anterior vitreous phase. It occurs due to defective neural crest migration during ocular development. Mutation in function of PITX2 gene on 4q25 and FOXC1 on 6p25 is the most common cause in the pathogenesis. These mutations produce a large spectrum of phenotypes of anterior segment dysgenesis, mild to severe form; with or without systemic manifestations [1-3]. The disease entities included in ASD are Axenfeld-Rieger syndrome (ARS), Peter’s anomaly, Iris hypoplasia, Primary congenital glaucoma and Aniridia. Posterior embryotoxon (prominent anterior displacement of Schwalbe’s line) is an important but not essential clinical feature of ARS. It can be seen in about 15% of normal eyes without any clinical significance [4]. Fifty percent of patients with ARS develop glaucoma [5,6]. Persistent pupillary membrane is a congenital remnant of the anterior tunica vasculosa lentis and is a very common congenital anomaly, seen in most of the normal new-born babies [7]. These persistent pupillary membranes do not affect vision, unless the pupillary opening is less than 1.5 mm in size. Management of persistent pupillary membrane depends on the severity of the membrane. Small persistent pupillary membranes can be managed conservatively as the vision is not obstructed. Severe persistent pupillary membranes may be excised surgically [8]. Rao A et al. have reviewed 56 cases of unclassified ARS and concluded that focal strands of anterior synechiae and variable corneal involvement may be present in atypical cases of ARS [9]. Only one case of ARS without posterior embryotoxon has been reported earlier [10].
Conclusion(s)
Axenfeld-Reiger’s syndrome is a rare form of ocular anterior segment dysgenesis. The current case represents an atypical isolated Reiger’s anomaly without posterior embryotoxon. This is a rare presentation and worth reporting as it exemplifies that posterior embryotoxon is not a major diagnostic criterion for Rieger’s anomaly.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Jul 15, 2020
Manual Googling: Oct 06, 2020
iThenticate Software: Dec 07, 2020 (11%)
[1]. Mears AJ, Jordan T, Mirzayans F, Dubois S, Kume T, Parlee M, Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomalyAm J Hum Genet 1998 63(5):1316-28.10.1086/3021099792859 [Google Scholar] [CrossRef] [PubMed]
[2]. Honkanen RA, Nishimura DY, Swiderski RE, Bennett SR, Hong S, Kwon YH, A Family with Axenfeld-Rieger syndrome and Peters anomaly caused by a point mutation (Phe112Ser) in the FOXC1 geneAm J Ophthalmol 2003 135(3):368-75.10.1016/S0002-9394(02)02061-5 [Google Scholar] [CrossRef]
[3]. Lehmann OJ, Ebenezer ND, Ekong R, Ocaka L, Mungall AJ, Fraser S, McGill JI, Ocular developmental abnormalities and glaucoma associated with interstitial 6p25 duplications and deletionsInvest Ophthalmol Vis Sci 2002 43(6):1843-49. [Google Scholar]
[4]. Malik S, Singh G, Gupta AK, Posterior EmbryotoxonIndian J Ophthalmol 1969 17:115-16. [Google Scholar]
[5]. Shields MB, Shields Textbook of Glaucoma 2011 6th edPhiladelphiaLippincott Williams & Wilkins:227-35. [Google Scholar]
[6]. Shields MB, Axenfeld-Rieger syndrome: A theory of mechanism and distinctions from the iridocorneal endothelial syndromeTrans Am Ophthalmol Soc 1983 81:736-84. [Google Scholar]
[7]. Tanzer DJ, McClatchey SK, Spontaneous hyphema secondary to a prominent pupillary membrane in a neonateJ Pediatr Ophthalmol Strabismus 1997 34:318-20.10.3928/0191-3913-19970901-15 [Google Scholar] [CrossRef]
[8]. Banigallapati S, Potti S, Marthala H, A rare case of persistent pupillary membrane: Case-based approach and managementIndian J Ophthalmol 2018 66(10):1480-83.10.4103/ijo.IJO_495_1830249846 [Google Scholar] [CrossRef] [PubMed]
[9]. Rao A, Padhy D, Sarangi S, Das G, Unclassified Axenfeld-Rieger Syndrome: A Case Series and Review of LiteratureSemin Ophthalmol 2018 33(3):300-07.10.1080/08820538.2016.120876727929720 [Google Scholar] [CrossRef] [PubMed]
[10]. Sim KT, Karri B, Kaye SB, Posterior embryotoxon may not be a forme fruste of Axenfeld-Rieger’s SyndromeJ AAPOS 2004 8(5):504-06.10.1016/j.jaapos.2004.06.01215492748 [Google Scholar] [CrossRef] [PubMed]