Membranoproliferative Glomerulonephritis is a glomerular change resulting from a wide range of disease sharing a common pathogenesis and thus, presented with large spectrum of clinical presentation starting from slowly progressive subnephrotic range proteinuria to nephritic syndrome and RPGN. Above that, it is one of the leading causes of End Stage Renal Disease (ESRD) all over the world and the recurrence rate following renal transplantation is very high [1]. Traditionally, MPGN was classified into three subclasses (type I, II, III) according to histopathological patterns as well as the location of electron dense deposits under Electron Microscope (EM). But this classification was not able to unveil the aetiologies behind the disease conditions and thus, fails to guide categorical opinion. Thereby, the proposed newer classification of MPGN based on Direct Immunofluorescence (DIF) divided it into immunoglobulin-mediated disease with overactivation of Classical Complement Pathway (CP) and non-immunoglobulin-mediated disease via activation of the Alternate Complement Pathway (AP) and lesion without any immune complex or complement deposition [2]. Very sparse literatures are available regarding the new classification [3,4]. In this study, the clinical data were correlated with the light microscopic and DIF findings to reanalyse the aetiological classification of MPGN and its prognostic outcomes as limited research works were available from West Bengal, India.
Materials and Methods
This was a retrospective, cohort study including the patients of all age groups who were diagnosed with MPGN pattern of injury after native kidney biopsies were done at Nephrology Department and reported in Department of Pathology of a tertiary care hospital in Kolkata, West Bengal for the time period of six months (February, 2018-August, 2018).
Inclusion criteria: The patients of all age group with MPGN pattern of injury were included.
Exclusion criteria: The patients with any other diagnosis, transplanted kidney, any primary neoplastic conditions and inconclusive results were excluded. Ethical committee approval and proper informed consent was taken (IEC no- IPGME&R/IEC/2016/580). Determination of sample size was done using www.openepi.com-Version 3 [5]. The sample size was calculated using following formula-
Sample size n={DEFF*Np(1-p)]/ [(d2/Z21-/2*(N-1)+p*(1-p)}
Where,
Population size (for finite population correction factor or FPC) (N)- 1000000, Hypothesised % frequency of outcome factor in the population (p) -8%+/-5, Confidence limits as % of 100 (absolute +/- %) (d)-5%, and Design effect (for cluster surveys- DEFF)-1. Considering 80% confidence interval, 49 patients were initially incorporated in the study. But considering previously mentioned exclusion criteria, two patients were subsequently eliminated from study population.
The following laboratory data were obtained from each of this study: complete blood count, blood glucose, urine analysis, serum urea, serum creatinine, Blood Urea Nitrogen (BUN), serum albumin, serum cholesterol, serum Hepatitis B surface antigen (HBs Ag), Anti-hepatitis C antibody (anti-HCV Ab), ASO titre, serum Anti-Nuclear Antibody (ANA), serum MPO ANCA, serum PR3 ANCA, serum dsDNA, serum C3 and C4 levels. Clinical parameters (age, sex, blood pressure, skin lesions, arthralgia, oedema, obesity) were also documented. Depending on the clinical presentation and laboratory parameters, all patients were categorised into four clinical syndromes [6]-
Occult nephritis: Haematuria and/or mild proteinuria (0.5 g/d < quantitative urinary protein < 1 g/d), or proteinuria (1 g/d ≤ quantitative urinary protein ≤ 3.5 g/d) without haematuria and eGFR ≥60 mL/min/1.73 m2.
Nephritic syndrome: Mild to moderate proteinuria (1 g/d ≤ urinary protein excretion ≤ 3.5 g/d) with haematuria, may be accompanied by oedema and hypertension and eGFR ≥60 mL/min/1.73 m2.
Nephrotic syndrome: High proteinuria (urinary protein >3.5 g/d), low serum albumin (<30 g/L), hyperlipidaemia, high degree of oedema and eGFR ≥60 mL/min/1.73 m2.
Renal failure: Decrease in Glomerular Filtration Rate (eGFR <60 mL/min/1.73 m2), may be accompanied by anaemia, hypertension or oedema.
Renal biopsies were done by tru-cut semiautomated renal biopsy gun. All specimens were examined by two nephropathologists at the institute using both light microscope and immunofluorescence microscope. H&E, PAS, silver methanamine, MT stained smears were prepared for light microscopy. Specimens for immunofluorescence microscopy were received in Michelle’s medium and were stained using FITC conjugated polyclonal rabbit anti-sera against human IgG, IgM, IgA, C3, C1q, kappa, lambda. Immunofluorescence findings were categorised from (+) to (++++) according to intensity. Location and pattern of staining were also documented. Control slides were also examined simultaneously. In light microscopy, diagnosis of MPGN was based on the presence of mesangial and endocapillary proliferation with irregular thickening and in some cases double contouring of the Glomerular Basement Membrane (GBM).
Statistical Analysis
The Kruskal–Wallis test was performed for comparisons between multiple groups. The χ2 test was analysed for categorical evaluation. Correlations were evaluated using Spearman’s rank correlation. A p<0.05 was considered as statistically significant. Statistical software (GRAPHPAD PRISM 5) was used for analysis.
Results
Analysis of Clinical and Biochemical Data
After the fulfillment of the inclusion and exclusion criteria, 47 cases were analysed in present study with mean age of 28.38±13.76 years. The male and female ratio was 1: 2.7. Other characteristics at the time of the diagnosis are listed in [Table/Fig-1]. Of significance are mean serum creatinine of 1.68±1.14 mg/dL, mean serum albumin of 2.70±0.91 mg/dL, mean proteinuria of 3.68±1.12 g/day, haematuria in 35 (74.46%) cases and hypertension in 4 (8.51%) cases.
Summary of clinical and biochemical parameters in the three groups on diagnosis.
| Parameters | Total cohort (N=47) | Immunoglobulin Positive (N=41) | C3 Positive (N=4) | IF Negetive (N=2) | p-value |
|---|
| Mean age (Years) | 28.38±13.76 (mean±SD) | 26.95±12.88 | 35.5±18.35 | 43.5±17.67 | 0.14 |
| Male:Female | 1: 2.7 | 9 : 32 | 2 : 2 | 1:1 | 0.34 |
| Serum creatinine (mg/dL) | 1.68±1.14 (mean±SD) | 1.62±0.92 | 1.49±0.72 | 3.05±0.21 | 0.09 |
| Proteinuria (g/day/1.73 m2) | 3.68±1.12 (mean±SD) | 3.71±1.87 | 3.8±1.39 | 1.75±0.21 | 0.33 |
| Serum albumin (g/dL) | 2.70±0.91 (mean±SD) | 3.14±1.35 | 3.0±0.95 | 2.35±0.21 | 0.70 |
| Serum C3 (mg/dL) | 58.45±42.6 (mean±SD) | 49.8±49.2 | 20.5±7.2 | 67.5±3.5 | 0.41 |
| Serum C4 (mg/dL) | 19.91±14.63 (mean±SD) | 14.31±14.92 | 25.95±14.19 | 35.0±4.24 | 0.07 |
| Arterial hypertension, n (%) | 4 (8.51) | 2 (4.87) | 0 (0.0) | 2 (100) | <0.0001* |
| Haematuria, n (%) | 35 (74.46) | 31 (75.60) | 2 (50) | 2 (100) | 0.37 |
N.B. The values for continuous variables are expressed as the mean±SD or as percentage. The Kruskal-Wallis test was performed for comparisons between multiple groups. The χ2 test was analysed for categorical evaluation. Correlations were evaluated using Spearman’s rank correlation. *p<0.05 was considered as significant; SD: Standard deviation
Groups According to Immunofluorescence Findings
Using the newer immunofluorescence-based MPGN classification system [2], there were 41 cases (87.23%) in the immunoglobulin positive group, four cases (8.51%) in the C3 positive group and two cases (4.25%) in the IF negative group. Clinical and biochemical parameters among the three groups are evaluated in [Table/Fig-2]. The mean age of the IF negative group was found to be higher than other two groups (immunoglobulin positive 26.95±12.88 years vs. C3 positive 35.5±18.35 years vs. IF negative 43.5±17.67 years). Serum creatinine was found to be high in all the groups on diagnosis but the IF negative group showed the maximum rise of serum creatinine level (immunoglobulin positive 1.62±0.92 mg/dL vs. C3 positive 1.49±0.72 mg/dL vs. IF negative 3.05±0.21 mg/dL). Both the immunoglobulin positive and C3 positive group presented with nephrotic range proteinuria whereas the IF negative group showed subnephrotic range proteinuria (immunoglobulin positive 3.71±1.87 g/day/1.73 m2 vs. C3 positive 3.8±1.39 g/day/1.73 m2 vs. IF negative 1.75±0.21 g/day/1.73 m2). Serum C3 value was low in C3 positive and immunoglobulin positive groups whereas IF negative group showed normal range of serum C3 level on diagnosis (immunoglobulin positive 49.8±49.2 mg/dL vs. C3 positive 20.5±7.2 mg/dL vs. IF negative 67.5±3.5 mg/dL). Serum C4 was found to be low only in immunoglobulin positive group (14.31±14.92 mg/dL). The IF negative group had 100% of cases with haematuria and hypertension [Table/Fig-1].
Systemic diseases associated with Membranoproliferative Glomerulonephritis (MPGN) on diagnosis.
| Disease | Immunoglobulin Positive (N=41) | C3 Positive (N=4) | IF Negative (N=2) | p-value |
|---|
| Infection, n (%) | 8 (19.51) | 1 (25) | 0 (0.0) | 0.74 |
| Autoimmune, n (%) | 29 (70.73) (ANA positive=26 cases; ANA negative=3 cases) | 0 (0.0) | 0 (0.0) | 0.0039* |
| Dysproteinemia, n (%) | 3 (21.43) | 0 (0.0) | 0 (0.0) | 0.79 |
| None, n (%) | 1 (2.44) | 3 (75) | 2 (100) | <0.0001* |
N.B. The Kruskal-Wallis test was performed for comparisons between multiple groups. The χ2 test was analysed for categorical evaluation. Correlations were evaluated using Spearman’s rank correlation. *p<0.05 was considered as significant; SD: Standard deviation
Infectious diseases predominate in the immunoglobulin positive group comprising eight of all cases in this group. Only one case in C3 positive group was associated with infections. In the immunoglobulin positive group, a clinical association with HCV infection (n=1), HBV infection (n=1) were found. Rest of the cases were associated with upper respiratory tract infection (n=6). Autoimmune disease was present in 29 patients of the immunoglobulin positive group [Table/Fig-2]: lupus nephritis-class IV defined as MPGN pattern of injury with full-house immune deposits in DIF and meeting the ACR (American College of Rheumatology) criteria for lupus was found in 26 cases [7]. Rest three cases were negative for serum ANA serology were diagnosed as Sjogren’s syndrome (n=1), mixed connective tissue disorders (n=2). Monoclonal gammopathy or dysproteinemia was found in three cases of immunoglobulin positive group: multiple myeloma (n=1), mantle cell leukaemia (n=1) and chronic lymphocytic leukaemia (n=1). Only one case in the immunoglobulin positive group was found to be not associated with any systemic disease whereas none of the cases in the IF negative group was found to be associated with any systemic disorders [Table/Fig-2].
A majority of the cases showed some degree of Tubular Atrophy and Interstitial Fibrosis (IFTA). Mild IFTA was seen in 33 cases (80.50%), two cases (50%) and one case (50%) of the cases in the immunoglobulin positive, C3 positive and IF negative groups, respectively. Approximately, seven cases (17.07%), two cases (50%) and one case (50%) of the cases in the immunoglobulin positive, C3 positive and IF negative groups revealed moderate Interstitial Fibrosis and Tubular Atrophy (IFTA). Severe degree of IFTA were found only in the immunoglobulin positive group. Fibrous crescents were also found only in three cases in the immunoglobulin positive group [Table/Fig-3].
Summary of light microscopy findings.
| Parameters | Autoimmune | Infection-associated | Dysproteinemia | C3 Nephropathy (n=4) | Pauci-Immune (n=2) | Overall (n=47) | p-value |
|---|
| Immunoglobin associated (n=41) |
|---|
| Neutrophils | 13/41 | 0/4 | 0/2 | 13/47 | 0.27 |
| Cellular crescent | 10/41 | 0/4 | 0/2 | 10/47 | 0.39 |
| Karryorhexis | 11/41 | 0/4 | 0/2 | 11/47 | 0.35 |
| Wire Loop | 10/41 | 0/4 | 0/2 | 10/47 | 0.39 |
| Interstitial fibrosis |
| <25% | 33/41 | 2/4 | 1/2 | 36/47 | 0.48 |
| 25-50% | 7/41 | 2/4 | 1/2 | 10/47 |
| >50% | 1/41 | 0/4 | 0/2 | 1/47 |
| Tubular atrophy |
| <25% | 29/41 | 2/4 | 1/2 | 32/47 | 0.66 |
| 25-50% | 9/41 | 2/4 | 1/2 | 12/47 |
| >50% | 3/41 | 0/4 | 0/2 | 3/47 |
| Fibrous crescent | 3/41 | 0/4 | 0/2 | 3/47 | 0.79 |
| Vascular involvement | 7/41 | 1/4 | 2/2 | 10/41 | 0.0196* |
N.B. The Kruskal-Wallis test was performed for comparisons between multiple groups. The χ2 test was analysed for categorical evaluation. Correlations were evaluated using Spearman’s rank correlation. *p<0.05 was considered as significant
The main findings in the vascular involvement were medial thickening and hyalinosis of the vessel wall and they were observed in seven cases (17.07%), one case (25%) and two cases (100%) of the cases in the immunoglobulin positive, C3 positive and IF negative groups, respectively, showing a significant statistical difference (p=0.0196) corresponding to more vascular involvement in the IF negative group [Table/Fig-4,5,6 and 7].
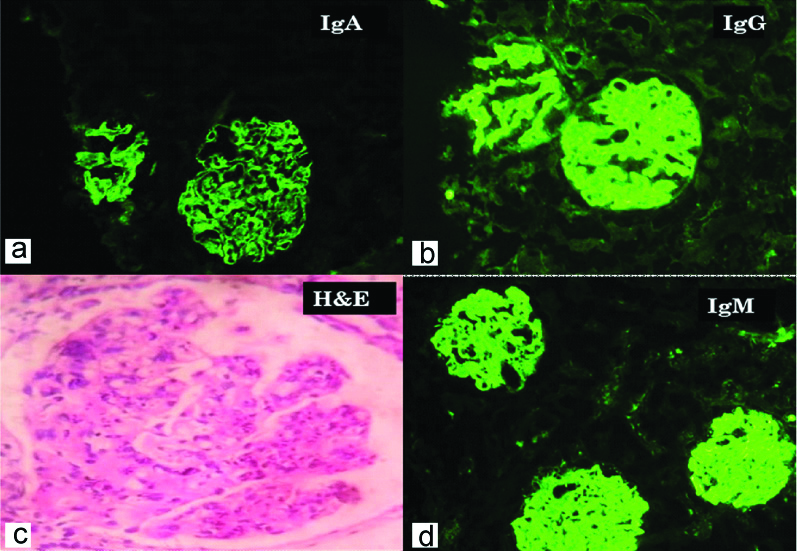
Case with full-house immunofluorescence positivity. a) Shows IgA immune deposits in mesangium and peripheral capillary wall (IgA, 100X); b) Shows IgG immune deposits in mesangium and peripheral capillary wall (IgG, 100X); c) Shows mesangial and endocapillary proliferation with wire loop lesion (H&E, 400X); d) Shows IgM immune deposits in mesangium and peripheral capillary wall (IgM, 100X).
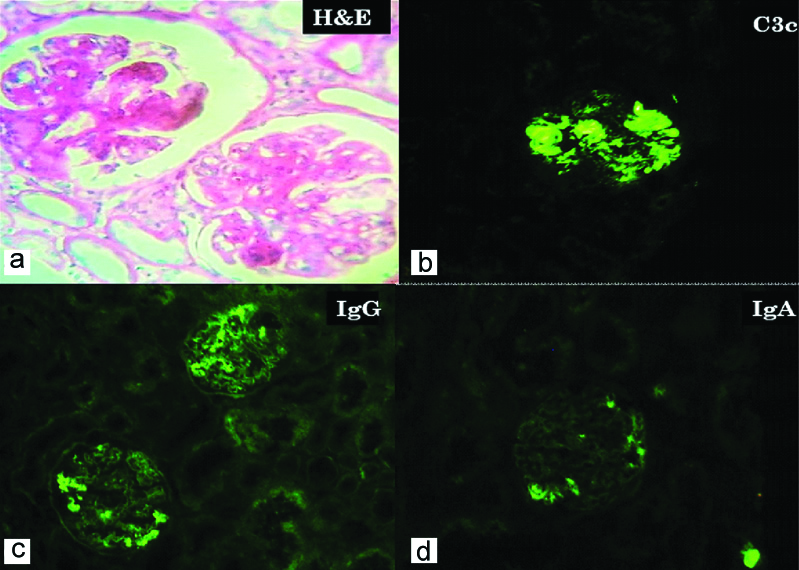
Case with predominant C3 deposition. a) Shows mesangial hypercellularity with accentuated lobular pattern (H&E, 100X); b) Immunofluorescence shows C3 immune deposits in mesangium. (C3c, 100X); c) Immunofluorescence shows trace and segmental IgG deposits (IgG, 100X); d) Shows trace IgA deposits (IgA, 100X).
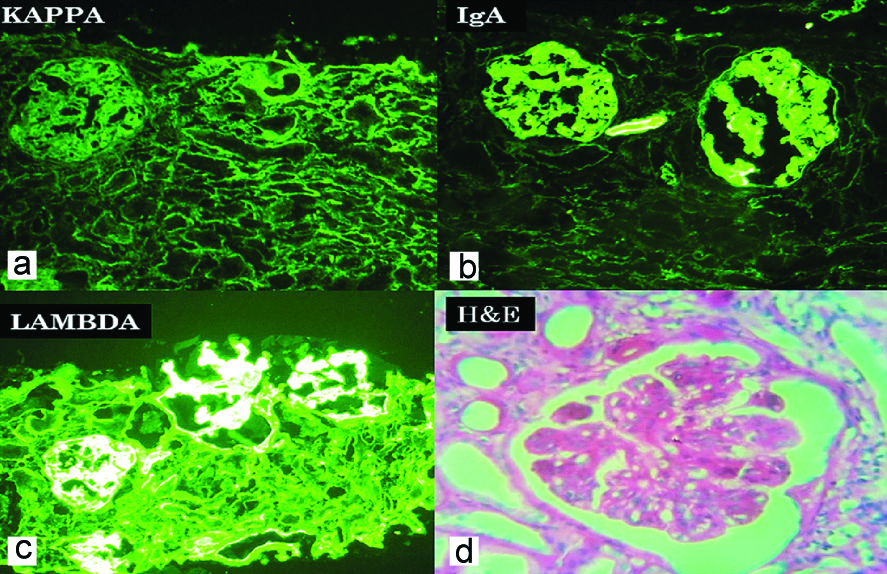
Immunofluorescence images of case with monoclonal immune deposit. a) (Kappa, 100X) shows no immune deposit (light chain restriction); b) (IgA, 100X) shows IgA immune deposits in mesangium; c) (Lambda, 100X) shows lambda light chain deposition in mesangium; d) (H&E, 400X) shows mesangial and endocapillary proliferation with wire loop lesion.
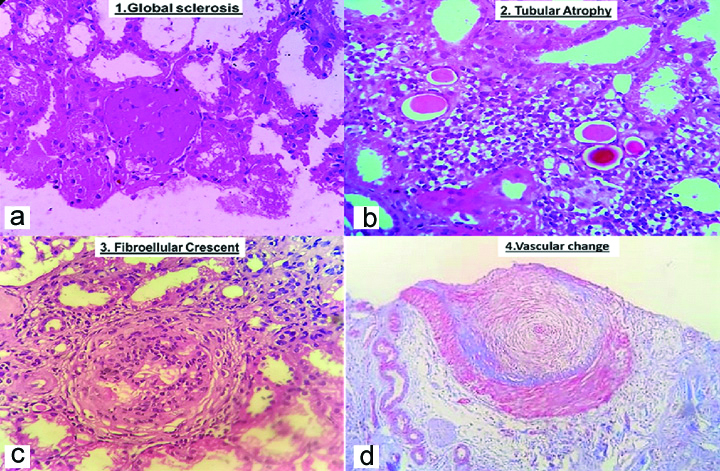
Showing chronic changes in all compartments of kidney. a) (H&E, 400X) shows global glomerulosclerosis; b) (H&E, 400X) shows tubular atrophy; c) (H&E, 400X) shows circumferential fibrocellular crescent; d) (H&E, 400X) shows mucoid intimal hyperplasia of the blood vessel.
IgG was found to be positive in 29 (70.73%) cases of immunoglobulin positive group (n=41) followed by IgM and IgA. ‘Full-house’ positivity was found in total nine cases. C3 was found positive in all cases of C3 positive group. Light chain restriction was found in all the cases which were labeled as Monoclonal Immune Deposit Disease (MIDD) among the immunoglobulin positive group (p≤0.001) [Table/Fig-8].
Summary of immunofluorescence findings.
| Parameters | Autoimmune | Infection-associated | Dysproteinemia | C3 Nephropathy (n=4) | Pauci-Immune (n=2) | Overall (n=47) | p-value |
|---|
| Immunoglobin-associated (n=41) |
|---|
| IgG | 29/41 | 0/4 | 0/2 | 29/47 | 0.0039* |
| IgM | 17/41 | 0/4 | 0/2 | 17/47 | 0.0477* |
| IgA | 11/41 | 0/4 | 0/2 | 11/47 | 0.34 |
| C3 | 9/41 | 4/4 | 0/2 | 13/47 | 0.0026* |
| C1q | 16/41 | 0/4 | 0/2 | 16/47 | 0.16 |
| Light chain restriction (only one light chain was present) | 0/29 | 0/8 | 3/4 | 0/4 | 0/2 | 3/47 | <0.001* |
N.B. The Kruskal-Wallis test was performed for comparisons between multiple groups. The χ2 test was analysed for categorical evaluation. Correlations were evaluated using Spearman’s rank correlation. *p<0.05 was considered as significant.
Discussion
Membranoproliferative Glomerulonephritis is renal disorder characterised by mesangial cell hypercellularity and architectural changes of GBM. Sethi S and Fervenza FC, depicted the difference in aetiopathogenesis between MPGN mediated by immunoglobulin deposition and MPGN resulting from the AP abnormality with only C3 deposition [2]. Another study emphasised that MPGN could be caused by Thrombotic Microangiopathy (TMA), where no immune deposits were found on IF [8].
Immunofluorescence assay revealed that vast majority (87.23%) of the MPGN cases in present study showed immunoglobulin deposition, whereas 8.51% cases showed C3 deposition without immunoglobulin and 4.25% cases showed no immunoglobulin or complement deposition.
Autoimmune diseases (61.70%) were found to be associated with majority of the cases. All of them were found in association with the immunoglobulin positive group (70.73%). Among 29 cases, 26 cases were found to be lupus nephritis defined as MPGN pattern of injury with full-house immune deposits in DIF and meeting the ACR criteria for lupus. Only three cases were found to be negative for serum ANA serology: one case was of Sjogren’s syndrome and the other two cases were diagnosed with rheumatoid arthritis and mixed connective tissue disorders. Zand L et al., found an association with autoimmune diseases other than lupus in 17 (5.5%) patients out of 308 with MPGN diagnosis, the most common being rheumatoid arthritis and Sjogren’s syndrome [9].
In the study, only nine cases were found to be associated with systemic infections. Among them eight cases (19.50%) were found in the immunoglobulin positive group and only one case (25%) was found in the C3 positive group. Most of the cases had an association with upper respiratory tract infection, but there were single cases with HBV and HCV infection as well. Proportionate distribution observed in both groups was attributed by sample size bias. Mostly in those cases, infection could have triggered dysregulation of complement cascade that is known as the basis of pathogenesis of C3 glomerulopathy. Woo SA et al., described complement mediated MPGN in 2 patients (4.3%) and immune-mediated MPGN in 39 patients (95.7%), out of 44 diagnosed with MPGN, not much different from the results that showed a predominance of the group with immune complex deposition [10].
MPGN relates to multiple myeloma, lymphoma and leukaemia whereas other paraproteinemias are rare [8,11]. Total 3 cases were associated with dysproteinemias: all of them were in the immunoglobulin positive group and immunofluorescence showed light chain restriction in all cases. Though Larsen CP et al., described MPGN associated with dysproteinemia patients with negative monoclonal immunoglobulin deposits that were unmasked only after performing immunofluorescence on formalin-fixed paraffin embedded tissue after protease digestion [12].
In relation to clinical and laboratory data, the immunoglobulin positive and C3 positive groups did not show any significant differences but the IF negative group had shown a little different presentation like maximum rise in serum creatinine compared to the other groups with subnephrotic range proteinuria, presence of hypertension and haematuria in all cases. Maximum vascular involvement was also found in IF negative group. MPGN with negative IF although not reported in the original classification systems, comprises 4.26% of the cases, and it should be seen as a group in which reparative process of TMA is involved, besides other cause [8].
Severe interstitial fibrosis, tubular atrophy and fibrous crescent were found only in immunoglobulin positive group which indicate that this group on follow-up might show chronicity or progression to ESRD. Nargund P et al., observed poorer prognosis in terms of chronic kidney disease and death in complement dominant group than immunoglobulin dominant MPGN [13]. Among the complement dominant group, proportion of ESRD was greater in Dense Deposit Disease (DDD) than C3 glomerulonephritis [14].
The complement system represents a primary defense modality. Its activation results in recruitment of neutrophils and pathogen elimination by apoptosis and cell lysis. The complement system is activated through three primary pathways-classic pathway, lectin pathway, and AP. The classic pathway and lectin pathway are triggered by Igs and bacterial carbohydrates, respectively, whereas the AP maintains a constant, spontaneous low level of activity with mechanisms that exist to maintain tight control, including activating and inhibitory proteins. Genetic mutations or acquired antibodies to these proteins result in dysregulation of the complement system.
In patients with C3 glomerulopathy, the estimation of serum C3, C4, C3NF, CFH, paraprotein and the screening for CFHR5 gene mutations were recommended [15].
This study attempts to show that MPGN is not a single disease entity rather this pattern of injury could be seen in broad spectrum of disease condition with different aetiopathogenesis.
Limitation(s)
Sample size was small and selection bias could not be avoided as it was an institution based study.
Conclusion(s)
Through this study, it has been established that various aetiologically diverse kidney diseases can manifest as MPGN both clinically and histomorphologically. Thus, MPGN can be regarded as a pattern of injury rather than a single disease entity. Critical evaluation of immunoflourescence finding using three heavy chains (IgG, IgA and IgM), two light chains (kappa and lambda) and two complements (C3c and C1q) helps in proper segregation and thus individualised management of this complex entity.