Adrenal tumours are relatively uncommon neoplasms, broadly divided into adrenal cortical and medullary tumours. They show bimodal age distribution. This is a retrospective case series of 36 cases of adrenal tumours over three years (September 2016- August 2019) in NRS Medical College Kolkata, West Bengal, India. Apart from Haematoxylin and Eosin (H&E) staining relevant Immunohistochemistry (IHC) (synaptophysin, Ki-67, Human Melanoma Black 45 (HMB 45) were done for proper histopathological diagnosis. Amongst 36 cases, 12 cases (34%) were myelolipoma, five cases (14%) were neuroblastoma, six cases (17%) of adrenocortical adenoma, two cases (06%) adrenocortical carcinoma, five cases (14%) pheochromocytoma, two cases (06%) mature teratoma, one case (03%) of epitheiloid PECOMA (Perivascular epithelioid cell tumour), one case haemorrhagic pseudocyst (03%) and two cases (06%) were metastasis from renal cell carcinoma. Eleven patients had specific hormonal symptoms. Bimodal age distribution with 10 cases in younger than 14 years, eight cases in more than 50 years and rest 18 cases were in range of 14-50 years. Overall male: female ratio is 5:4 but myelolipoma was common in female. We have encountered few rare entities-Epithelioid PECOMA (angiomyolipoma), Pheochromocytoma with Von Hippel-Lindau (VHL), Receptor for Neurotrophic Factor (RET) Mutation and uncommon presentation with Cushing’s Syndrome, one Adrenal Oncocytoma and two Mature Teratoma.
Introduction
Adrenal glands are composed of mesodermally derived cortex (90% of total weight) and neuroectodermally derived medulla. Adrenocortical tumours may be hormone secreting or non-functioning and are divided into adrenocortical adenoma and carcinoma. Adrenal medullary tumours are neuroblastic tumours in paediatric age group and pheochromocytoma in both adult and childhood. Other adrenal mass lesions includes cyst, pseudocyst, myelolipoma, teratoma, stromal tumours, angiomyolipoma etc. Among these adrenal myelolipoma is quite common, sometimes presenting as incidetaloma. Adrenal incidentaloma exist in about 2-3% of general population and there incidence increases with age (1% at 40 years and 7% at 70 years) [1]. Some of the pheochromocytomas are associated with familial genetic disorders like MEN 2A, 2B, VHL mutation, Von Recklinghausen disease [1]. Considering rarity and various pathological entities of adrenal tumours, different clinical symptoms, relevant biochemical findings, authors decided to share their experience of 36 adrenal tumours in a Tertiary Health Institution in different age groups.
Case Series
This was a three years retrospective case series (September 2016 to August 2019) with 36 cases of adrenal tumours in different age groups including paediatric patients, done in the Department of Pathology and Urology, NRS Medical College and Hospital, Kolkata, West Bengal, India. Clinical and investigational data were collected from hospital records. All histopathological reports and gross features were received from pathology record register. Along with H&E stained sections, special stains like Reticulin stain were also examined. In few selected cases IHC with Ki-67, HMB 45, Synaptophysin staining were performed when there was diagnostic dilemma. Distribution of pathological diagnosis of 36 cases of adrenal masses are shown in [Table/Fig-1] and distribution of cases according to the symptoms are shown in [Table/Fig-2]. Among 36 cases of adrenal lesions, 10 were malignant (27.78%) and 26 cases (72.22%) were benign. Both benign and malignant tumours were slightly more common in male with exception of myelolipoma. Most of the malignant tumours were seen in <14 years and 51-60 years age group. In 20 cases left, adrenal and 16 cases right adrenal were involved [Table/Fig-3].
Distribution of cases according to age groups (n=36).
| Cases | Paediatric age group (n=10) | Adult age group (n=26) | Total and percentage (n=36) |
|---|
| Neuroblastoma | 5 | 0 | 5 (13.89%) |
| Adrenal myelolipoma | o | 12 | 12 (33.33%) |
| Adrenal adenoma | 1 | 5 | 6 (16.67%) |
| Adrenocortical carcinoma | 1 | 1 | 2 (5.56%) |
| Pheochromocytoma | 2 | 3 | 5 (13.89%) |
| Teratoma | 1 | 1 | 2 (5.56%) |
| Angiomyolipoma | 0 | 1 | 1 (2.78%) |
| Pseudocyst | 0 | 1 | 1 (2.78%) |
| Metastasis | 0 | 2 | 2 (5.56%) |
Distribution of cases according to presenting symptoms (n=36).
| Presenting symptoms | No of cases |
|---|
| Incidental finding | 9 |
| Abdominal pain | 11 |
| Lump abdomen | 4 |
| Hypertension and headache | 5 |
| Cushing syndrome | 3 |
| Conn’s syndrome | 2 |
| Hirsutism and precocious puberty | 2 |
Distribution of benign and malignant cases according to age, sex and laterality (n=36).
| Cases | | Benign (n=26) | Malignant (n=10) |
|---|
| Sex | Male | 14 | 06 |
| Female | 12 | 04 |
| Age (years) | <14 | 04 | 06 |
| 14-30 | 03 | 00 |
| 31-40 | 08 | 00 |
| 41-50 | 06 | 01 |
| 51-60 | 05 | 03 |
| Laterality | Left | 14 | 06 |
| Right | 12 | 04 |
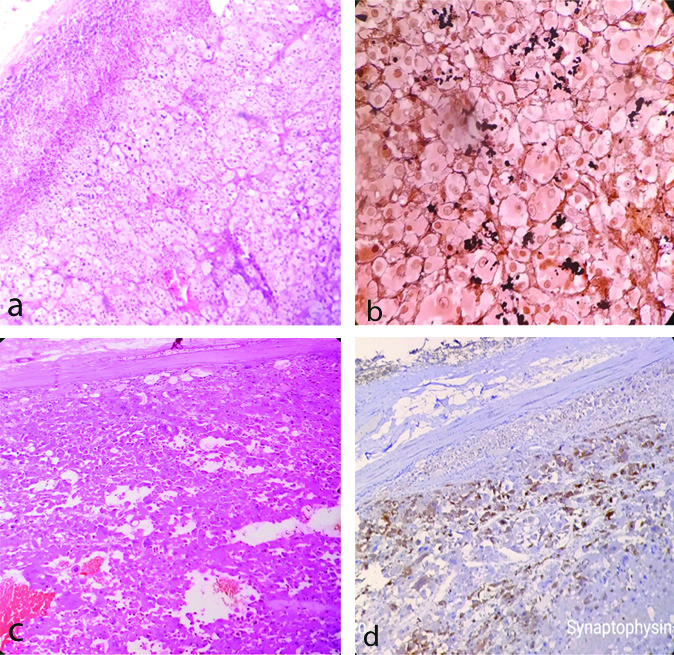
Among six cases of adrenal adenoma [Table/Fig-4a], five presented with hormonal symptoms. Two cases had features of Conn Syndrome (hypernatremia, hypokalaemia and hypertension). One female patient of such case also gave history of amenorrhoea. These cases showed raised aldosterone level. One case, a six-year old child presented with precocious puberty and enlargement of testis along with increased testosterone level. Two cases showed Cushing’s Syndrome with raised cortisol level. Special stain with reticulin showed typical nesting pattern [Table/Fig-4b]. One adrenal oncocytoma [Table/Fig-4c] presented as incidentaloma without any hormonal manifestations. IHC with synaptophysin positivity confirmed the diagnosis [Table/Fig-4d].
a) Adrenal adenoma (H&E X400) -showing nested pattern of growth composed of lipid rich cells and compressed adrenal at the periphery. b) Adrenal adenoma (Reticulin stain X400) -showing reticulin fibres highlighting nested pattern. c) Adrenal Oncocytoma (H&E X400) -showing large polygonal cells with intensely eosinophilic cytoplasm arranged in sheets and nest. d: Microphotograph of adrenal oncocytoma (IHC X400) showing synaptophysin positivity in tumor cells.
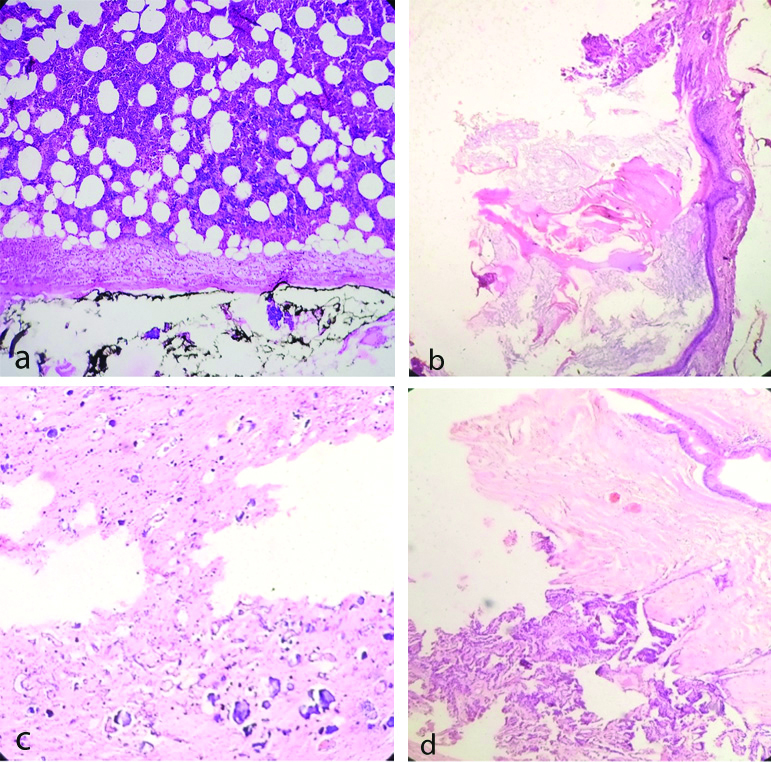
Twelve adrenal myelolipoma [Table/Fig-5a] showed haematopoetic and adipocytic cells. There were two cases of adrenal teratoma one in adult [Table/Fig-5b] and other in paediatric age. Both the cases showed histological features of mature teratoma. Paediatric case was misdiagnosed as myelolipoma radiologically due to high fat content. Histologically there were extensive chicken wire calcifications [Table/Fig-5c] in matured glial component and choroid plexus [Table/Fig-5d] which were rare findings. In both cases, normal adrenal tissue was completely replaced by the teratoma.
a) Adrenal myelolipoma (H&E X400) showing haematopoietic cells admixed with adipocytes. b) Adrenal teratoma in adult (H&E X400) showing mature teratoma containing skin adnexal structures and keratinous material. c) Adrenal teratoma in child (H&E X400) showing foci of calcification, intervening glial element. d) Same pediatric teratoma-showing choroid plexus, glandular element and glial tissue.
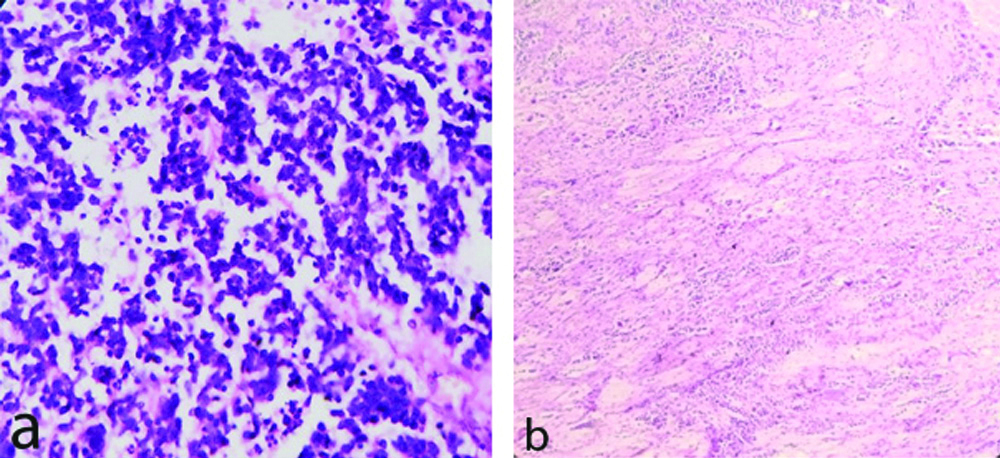
Among five neuroblastic tumours, four were poorly differentiated neuroblastoma [Table/Fig-6a], other one was ganglioneuroblastoma [Table/Fig-6b]. Two cases presented with cervical lymphnode metastasis i.e. stage IV disease. All cases showed raised VMA level in urine.
a: Neuroblastoma-Poorly differentiated (H&E X400) showing predominantly neuroblastic cells in groups, nests and rosette formation, neuropil in the back ground without schwannian stroma. b) Ganglioneuroblastoma-intermixed (schwannian stroma rich){ H&E X100} showing microscopic foci of neuroblastic cells and fair number of ganglion cells in an expanding schwannian stroma.
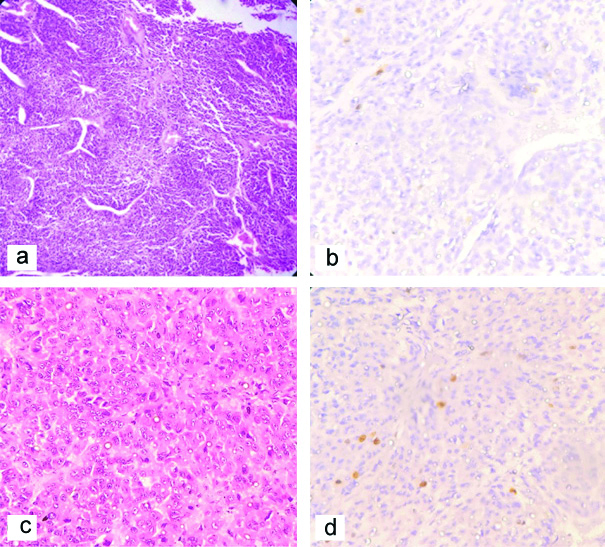
In this case series, five cases of Pheochromocytoma were found of which one patient aged 12 years showed bilateral involvement and VHL positive. One case, 45 years female showed RET mutation and was associated with medullary carcinoma thyroid. All cases presented with persistent or paroxysmal hypertension and headache. They had increased VMA level in urine and metanephrine levels in serum. Four cases showed classical histological feature of pheochromocytoma [Table/Fig-7a]. Bilateral Pheochromocytoma showed occasional nuclear pleomorphism, foci of necrosis and degenerative calcification but infrequent mitosis along with Ki67 index 1% [Table/Fig-7b]. Interestingly one case, 4.5 year female child presented with cushing syndrome, increased cortisol level which was not supressed by both low and high dose of dexamethasone. She had normal VMA level in urine. This case histologically showed focal diffuse pattern of growth [Table/Fig-7c], mild to moderate nuclear atypia, capsular infiltration but infrequent mitosis and Ki67 index 3% [Table/Fig-7d]. There was no associated adrenocortical hyperplasia. We diagnosed this case of pheochromocytoma having ectopic Adrenocorticotropic hormone (ACTH) secretion by tumour cells.
a) Pheochromocytoma (H&E X100) showing polygonal cells with granular eosinophilc cytoplasm round nucleus vescicular chromatin arranged in typical Zellballen Pattern. b) Pheocromocytoma (x400) IHC stain by Ki-67 in showing Ki-67 index <2%. c) Pheochromocytoma (H&E X400)-diffuse pattern of growth. d) Pheochromocytoma (IHC X400) showing Ki-67 immunostain having Ki 67 index 3%.
Among two cases adrenocortical carcinoma, one adult case (45 years male) did not show any hormonal manifestations. Other patient (three years female child) presented with hirsutism, hypertrophied clitoris (raised androgen). Both cases showed distant metastasis. Histologically they had high nuclear grade, increased mitosis (>5/50 hpf) [Table/Fig-8a] diffuse architecture in reticulin stain, capsular invasion thus, fulfilling Weiss criteria [2]. IHC showed Ki 67 index >5% [Table/Fig-8b].
a) Adrenocortical carcinoma (H&E X400)-showing large polygonal cells with nuclear atypia, diffuse pattern of growth and frequent mitoses. b) Adrenocortical carcinoma IHC staining by Ki67 (x400)-showing Ki-67 index >5%.
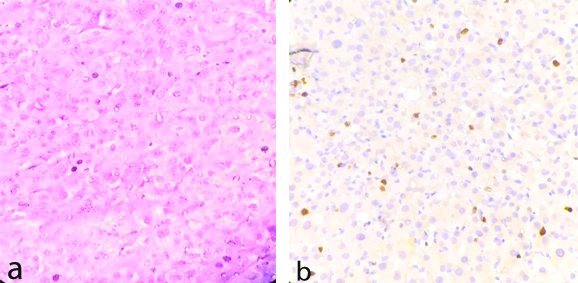
One rare case of adrenal angiomyolipoma (epithelioid variety) in a 52 years/male presented with large abdominal lump. Histologically it showed large polygonal cells with eosinophilic cytoplasm, atypical nuclei and prominent nucleoli, mitoses (>2/50 HPF) and foci of haemorrhage and necrosis [Table/Fig-9a]. This case was confirmed by IHC examination (HMB 45) [Table/Fig-9b]. Two cases of adrenal gland resected along with radical nephrectomy showed metastatic deposit from clear cell renal cell carcinoma [Table/Fig-10a]. One case of cystic lesion showed haemorrhagic material without any obvious cyst lining and thus diagnosed as adrenal pseudocyst [Table/Fig-10b].
a) Epithelioid Angiomyolipoma /PECOMA (H&EX400)-showing epithelioid cells with clear to granular eosinophilic cytoplasm and round uniform nucleus, few mitoses arranged in sheets. b) Epithelioid Angiomyolipoma/PECOMA IHC Staining by HMB 45-showing positive in tumor cells.
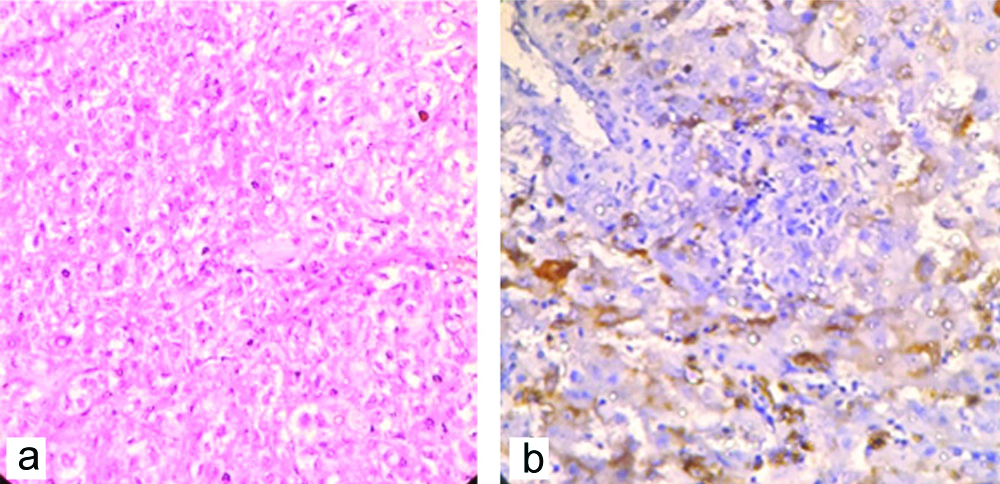
a) Adrenal gland metastasis-from clear cell carcinoma of kidney (H&E X100). b) Adrenal pseudocyst (H&E X100)-showing a cystic structure without any epithelial lining and adjacent normal adrenal tissue.
Discussion
Adrenocortical neoplasms may be adenoma and carcinoma. Adrenal adenomas are the most common neoplasm arising-adrenal gland [1]. These lesions may be found in approximately 6% patients in autopsy series. Adenoma may presents with hormonal symptoms in 7.1% of cases [1]. Adenomas are yellowish colour on gross pathological examination due to presence of abundant lipid. Histologically sometimes there may be difficulties in differentiating it from adrenocortical carcinoma [2]. Criteria proposed by Weiss and IHC are used to differentiate both of them. In the present case series the authors found six cases of adenomas -five in adult and one in pediatric population. Out of six cases of adenoma, total five cases presented with features of hormone hypersecretion-two cases presented with features of cortisol hypersecretion, two cases of aldosterone hypesecretion and one case of androgen hypersecretion. High incidence of metabolically active adrenoma in this case series was probably due to indications of operation of adrenal adenoma is either big size or metabolic activities in Tertiary Referral Centre. Adrenal oncocytoma are rare with approximately only 50 cases been reported in literature [1]. Histologically they are rich in mitochondria, large eosinophilic cells with abundant granulations. Pathogenesis is still not clear but more commonly seen in female (2.5 times more common than male) and left side is involved more commonly. About 90% cases are metabolically inactive [1]. In present series also, metabolically inactive one oncocytoma was found which was confirmed by synaptophysin (IHC).
Adrenal myelolipomas are rare, benign, metabolically inactive lesion involving less than 0.1% of autopsy series [1]. These lesions arise from clonal stem cell proliferation and are composed of an unusual mixture of mature adipose tissue and relatively normal haematopoietic elements. Presence of macroscopic fat in adrenal mass is virtually diagnostic of myelolipoma. There is a controversy whether myelolipoma to be regarded as neoplasm or reactive process. Bishop E et al., shows non-random X chromosome inactivation in both fat and haematopoietic element from female patient which supports its clonal origin [3]. In this case series, 12 cases of myelolipoma were found, mostly in females .
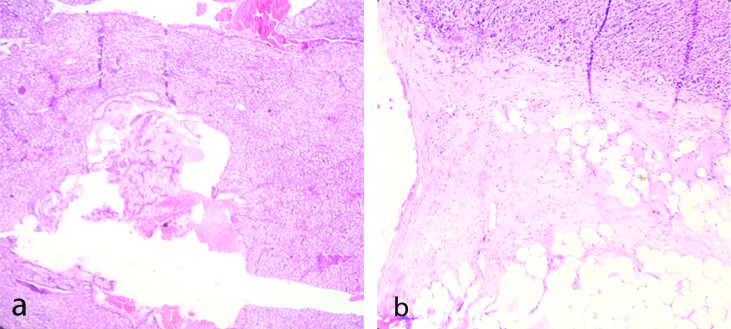
Adrenal cysts have been found in 0.06 to 0.18% population in autopsy series [1]. Four histologic types of adrenal cysts have been described namely pseudocyst, epithelial cyst, endothelial cyst and parasitic cyst. Pseudocysts are most common variety of adrenal cyst and they do not posses a cellular lining and are believed to be the result of previous intraadrenal haemorrhage or infarction. Upto 7% adrenal pseudocyst can be malignant. In this case series one case of pseudocyst was found.
Adrenocortical carcinoma is a rare malignancy with an incidence of 0.5 to 2 per million [1]. It has bimodal age distribution with two peaks (children-1st decade and adult 4th decades) [1]. Carcinoma should be diagnosed by using modified Weiss criteria [2]. In paediatric adrenocortical carcinoma, Weiss criteria is less predictive of an aggressive outcome. Cagle PT et al., stated weight of the tumour >500 gm as single useful determinant factor [4]. Recently Dehler LP and Hill A reviewed 39 adrenocortical neoplasms and suggested tumour weighing <200 gm and confined within adrenal are in low risk category [5]. Among two cases of adrenocortical carcinoma in this series, one case presented with increased testosterone level (paediatric), other without any hormonal symptoms.
Adrenal medullary tumours are neuroblastoma and ganglioneuroblasoma in paediatric age group and pheochromocytoma in adult. About 40% of neuroblastoma are involving <1 year age and 90% are diagnosed before five year [6]. International Neuroblastoma Pathology Committee (INPC) divided neuroblastic tumours into neuroblatoma, ganglioneuroblastoma and ganglioneuroma according to presence of ganglion cells, schwannian stroma and blastic components [7,8], in present series five cases of neuroblastoma were found.
Pheochromocytomas are catecholamine secreting tumour of adrenal medulla with incidence of 1-2 cases per lakh populations [1]. Amongst incidentally discovered adrenal masses, 5% will have pheochromocytoma [1]. Patients mostly presented with intermittent episodes of hypertention due to excessive production of catecholamines by tumour cells. Rarely it may be associated with paraneoplastic syndromes like Cushing syndrome, diarrhoea, hypokalemia, polycythemia etc [9-11]. Here we experienced a case of pediatric pheocromocytoma presenting with cushing syndrome.
Lam KY and Lo CY stated that adrenal gland is the 4th most common site of metatsasis; primary sites being breast, lung, kidney etc., [12]. In this case series two adrenal metastasis from renal cell carcinoma were found.
Adrenal teratomas are extremely rare tumour of adrenal gland (0.13% of all adrenal tumour) [13]. They are more common in adults. In this case series the authors found a paediatric adrenal teratoma which is extremely rare. Histopathologically they are mature teratoma with favourable outcome.
Angiomyolipoma is messenchymal tumour composed of adipose tissues, spindle cells, epithelioid smooth muscle cells and thick walled blood vessels. It belongs to a family of Perivascular epithelioid cell tumours (PECOMAs). Epithelioid angiomyolipoma is a rare variant consisting of Atleast 80% epithelioid cells [14] and can be of malignant potential.
Adrenal tumours arising from cortex or medulla show not only different histological entities but also varied presentations involving both adult and paediatric age groups. Tumour without hormonal manifestations usually present late with vague symptoms.
Conclusion(s)
In this retrospective case series of 36 adrenal tumours, 10 were malignant and rest were benign. Malignant tumours had a bimodal peak age <14 yr and 50-60 yrs. As a whole adrenal tumours had male predominance except myelolipoma. The present authors got few rare entities including epithelioid PECOMA (angiomyolipoma), pheochromocytoma with VHL, RET mutation and uncommon presentation with cushing syndrome, adrenal oncocytoma and mature teratoma. These patients may present in various ways from asymptomatic to symptoms of local features (pain, lump) or metabolic/hormonal features (headache, hypertension, amenorrhoea, precocious puberty). Histopathological examination with H&E stain are effective in diagnosis in most cases however in some selected cases special staining (Reticulin, PAS) or IHC may be needed for accurate diagnosis.
[1]. Alan J Wein, Louis R Kavoussi, Alan W Partin, Campbell Walsh Urology 2016 11th edPhiladelphia, PAElsevierChapter 65: Pathophysiology, evaluation and medical management of adrenal disorders: p. 1545-47 [Google Scholar]
[2]. Weiss LM, Comparative histologic study of 43 metastasizing and non metastasizing adrenocortical tumorsAm J Surg Pathol1984(8):163-69.10.1097/00000478-198403000-000016703192 [Google Scholar] [CrossRef] [PubMed]
[3]. Bishop E, Eble JN, Cheng L, Adrenal myelolipoma show non-random X chromosome inactivation in haematopoetic elements and fat: Support for a clonal origin of myelolipomaAm J Pathol 2006 30:838-34.10.1097/01.pas.0000202044.05333.1716819325 [Google Scholar] [CrossRef] [PubMed]
[4]. Cagle PT, Hough A, Pyeher J, Comparisons of adrenocortical tumors in children and adultsCancer 1986 57:2235-37.10.1002/1097-0142(19860601)57:11<2235::AID-CNCR2820571127>3.0.CO;2-O [Google Scholar] [CrossRef]
[5]. Dehler LP, Hill A, Adrenocortical neoplasm in children: Why so many carcinomas and yet so many survivorsPediatr Develop Pathol 2009 12:284-91.10.2350/08-06-0489.119326954 [Google Scholar] [CrossRef] [PubMed]
[6]. Gurney JG, Ross JA, Wall DA, Bleyer WA, Severson RK, Robison LL, Infant cancer in US: Histology specific incidence and trends, 1973-1992J Pediatr Haematol Oncol 1997 19(5):428-32.10.1097/00043426-199709000-000049329464 [Google Scholar] [CrossRef] [PubMed]
[7]. Shimada H, Ambros IM, Dehner LP, Terminology and morphologic criteria of neuroblastic tumors: Recommendations by International Neuroblasoma Pathology CommitteeCancer 1999 86:349-63.10.1002/(SICI)1097-0142(19990715)86:2<349::AID-CNCR20>3.0.CO;2-Y [Google Scholar] [CrossRef]
[8]. Broduer G M, Seegar R C, Brrett A, International criteria for diagnosis, staging and response to treatment in patients with neuroblastomaJ Clin Oncol 1988 6:1874-81.10.1200/JCO.1988.6.12.18743199170 [Google Scholar] [CrossRef] [PubMed]
[9]. Nijhoff MF, Dekkars OM, Vleming LJ, ACTH producing pheochromocytoma: Clinical consideration and concise review of the literatureEur J Intern Med 2009 20:682-85.10.1016/j.ejim.2009.08.00219818286 [Google Scholar] [CrossRef] [PubMed]
[10]. Loehry CA, Kingham JG, Whorwell PJ, Watery diarrhoea and hypokalemia associated with pheochromocytomaPost grad Med J 1975 51:416-19.10.1136/pgmj.51.596.416175362 [Google Scholar] [CrossRef] [PubMed]
[11]. Pacak K, Jochmanova I, Prodanov I, New syndrome of paraganglioma and somatostatinoma associated with polycythemiaJ Clin Oncol 2013 31:1690-98.10.1200/JCO.2012.47.191223509317 [Google Scholar] [CrossRef] [PubMed]
[12]. Lam KY, Lo CY, Metastatic tumors of adrenal glands: A 30 year experience in a teaching hospitalClin Endocrinol (oxf) 2002 56:95-101.10.1046/j.0300-0664.2001.01435.x11849252 [Google Scholar] [CrossRef] [PubMed]
[13]. Narla SL, Jacob S, Kurian A, Parameswaran A, Primary mature cystic teratoma with carcinoid mimicking adrenal tumor: Report of a rare association and review of literatureIndian J Pathol Microbiol 2016 59:200-02.10.4103/0377-4929.18201227166041 [Google Scholar] [CrossRef] [PubMed]
[14]. Godara R, Vashist MG, Singla SL, Garg P, Sen J, Mathur SK, Adrenal angiomyolipoma: A rare entityIndian J Urol 2007 23(3):319-20.10.4103/0970-1591.3373419718340 [Google Scholar] [CrossRef] [PubMed]