Paediatric Embryonal Tumours in Multilayered Rosettes Presenting as a Low Grade Glioma- An Unusual Case Report
Sibhi Ganapathy1, Nikunj Godhani2
1 Associate Consultant, Department of Neurosurgery and Spine Surgery, Manipal Hospital Whitefield, Bengaluru, Karnataka, India.
2 Associate Consultant, Department of Neurosurgery and Spine Surgery, Manipal Hospital Whitefield, Bengaluru, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sibhi Ganapathy, 13 Waterwoods, Varthur Road Whitefield, Bangalore, Karnataka, India.
E-mail: sibhig09@gmail.com
Paediatric Embryonal Tumours in Multilayered Rosettes (ETMR) are rare aggressive tumours with poor survival statistics, defined in 2016 by World Health Organisation (WHO) classification of brain tumours. The tumours have a characteristic radiological appearance on Magnetic Resonance Imaging (MRI) of the brain, which is easily decipherable. This combined with a clinical picture of raised intracranial pressure symptoms, seizures and rapidly progressive new onset neurological deficits make the diagnosis fairly obvious. The final confirmation of the diagnosis is done by immunohistochemical analysis of the C19Myc gene alteration. Rarely, certain radiological presentations are uncharacteristic and resemble other more benign pathologies with overlapping clinical presentations. This can be misleading, as ETMRs require aggressive surgery followed by adjuvant chemotherapy and radiation to ensure best possible survival. We present such a case report of what appeared to be a low-grade glioma in the frontal lobe. This tumour presented with one episode of generalised tonic clonic seizures not unusual as a presenting complaint in low-grade gliomas per se. Surgical debulking under ultrasonic guidance was done and the specimen was sent to histopathological analysis. The histopathological analysis showed a surprise ETMR diagnosis which was sent for confirmation to two other centers. This case report highlights the need to keep ETMRs as a rare differential diagnosis for even low-grade gliomas of the brain, thereby allowing accurate prognostication only after histopathological and immunohistochemical assessment. We present a brief literature review on unusual presentations of ETMRs reported in literature to further illustrate the chimeric nature of this rare disease.
Neuroectodermal tumours, Poor prognosis, Seizures
Case Report
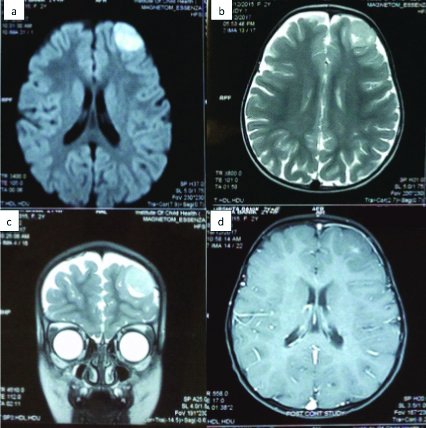
A two-year-old female child presented to our clinic two days after an episode of non-febrile, generalised Tonic Clonic Seizures. The seizure profile was as follows: Aura was absent. The ictus consisted of tonic-clonic movements of bilateral upper and lower limbs, coupled with tongue bite and incontinence. The ictus lasted for 1-2 minutes. In the post-ictal state, the child was confused and drowsy for 15 minutes before regaining normal consciousness. The child was immediately evaluated for the cause of this new onset seizure. The MRI of the child showed a left frontal lesion with minimal enhancement of contrast. The lesion was well-demarcated with a uniform consistency. No cystic or hyperintense regions were seen. The surrounding brain appeared normal with no oedema. There was no mass effect on the surrounding brain seen [Table/Fig-1a-d]. The possible differential diagnosis considered were Ganglioglioma, or Pleomorphic Xanthoastrocytoma (PXA). Based on this, she was advised surgery. She was taken up for a left frontal craniotomy, and navigation and ultrasound guided excision of the lesion under general anaesthesia.
a) Showing Axial DWI image; b) Shwoing axial T2; c) Coronal T2W; d) Axial T1W with contrast image showing the frontal polar well circumscribed lesion with almost no perilesional reaction suggestive of a low-grade glioma.
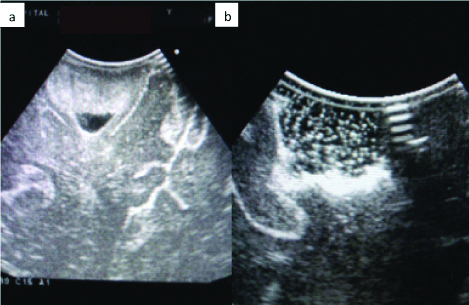
Total excision of the affected lesion was confirmed by on-table neuro-ultrasound of the tumour bed [Table/Fig-2]. Ultrasonic localisation was essential as tumour tissue was indistinguishable from the normal brain parenchyma. No oedema or increased vascularity was seen in comparison to the normal tissue. Post-excision the child was observed for three days in hospital and then, eventually discharged on antiepileptics to review after a week in order to obtain the histopathological diagnosis and thereby, plan adjuvant therapy if required. No decongestants were added as there was absolutely no oedema and postoperative Computerised Tomography (CT) scans of the brain showed no such features.
a) Showing tumour seen with on-table ultrasound; b) Shows the same image postoperative where the tumour is completely removed and replaced by the brownian movement of saline.
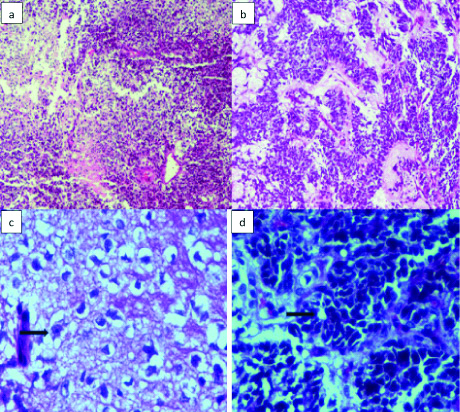
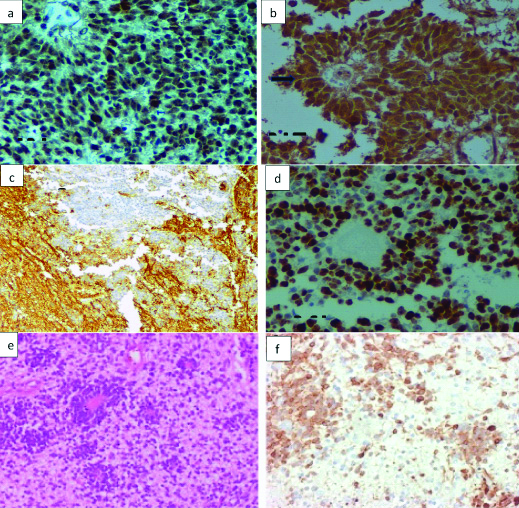
Histopathological analysis showed a biphasic cellular tumour which is composed of monomorphic cells arranged in sheets and papillary pattern interspersed with foci of small embryonal cells with hyperchromatic nuclei, scant cytoplasm, brisk mitotic activity, apoptotic bodies and necrosis [Table/Fig-3a]. The cells were seen on a background of fibrillary stroma, with ganglionic cells also were seen [Table/Fig-3b]. The cells showed focal perivascular resetting, occasional focus of palisading cells with papillary like configuration occasionally [Table/Fig-3c]. The presence of multilayered rosettes is unequivocal [Table/Fig-3d]. At this juncture, the radiological appearance of a low-grade glioma became incompatible with the histological appearance. The presence of rosettes as well as the abundance of neutropil pointed towards a neuronal lesion as compared to gliomas. A ganglioneuroblastoma was first considered before immunohistochemical staining was employed. Another possibility was Atypical Teratoid Rhabdoid Tumour (ATRT) which is rare but possible. The absence of rhabdoid features and the staining pattern as described made this impossible to defend. Immunohistochemical stains done showed Integrase Interacter-1 (INI-1) retained expression [Table/Fig-4a], Neuronal Nuclei (Neu-N) positive confirming the presence of embryonic component [Table/Fig-4b], synaptophysin positive for the fibrillary stroma alone [Table/Fig-4c], Glial Fibrillary Astrocyte Protein (GFAP)-negative indicating no proliferative glial component and a Mind Bomb E3 Ubiquitin Protein Ligase 1 (MIB-1) index of 80% indicating WHO grade 4 tumour [Table/Fig-4d]. Lin28 analog A (Lin28-A) was done as a surrogate marker for C19Myc detection. It was strongly positive indicating C19Myc amplification [Table/Fig-4f]. The immunohistochemical differential diagnosis can be made as described in [Table/Fig-5]. Differentiation of various similar differentials based on immunohistochemical markers are listed in [Table/Fig-6] [1-8].
a) Showed a biphasic cellular tumour composed of monomorphic cells arranged in sheets and papillary pattern interspersed with foci of small embryonal cells with hyperchromatic nuclei, scant cytoplasm, brisk mitotic activity, apoptotic bodies and necrosis (X40 H&E); b) The cells were seen on a background of fibrillary stroma, with ganglionic cells also were seen (X100 H&E); c) The cells show focal perivascular resetting, occasional focus of palisading cells with papillary like configuration occasionally (X400 H&E); d) The presence of multilayered rosettes is unequivocal (X400 H&E).
a) Immunohistochemistry showing INI-1 retained expression (X100 INI-1); b) Neu-N positive confirming the presence of embryonic component (arrow mark) (X400 Neu-N); c) Synaptophysin positive for the fibrillary stroma alone (X40 Synaptophysin); d) GFAP-negative indicating no proliferative glial component and a MIB-1 index of 80% indicating WHO grade 4 tumour (X100 MIB-1); e) Shows a normal eosin and hematoxylin stained film for comparison (X40 H&E); f) LIN28A was done as a surrogate marker for C19Myc detection. It was strongly positive indicating C19Myc amplification (X40 LIN28A).
Biomarkers to diagnose differentials masquerading as ETMRs.
| Biomarker | Diagnosis |
|---|
| C19MC (LIN28A expression) | ETMR |
| SMARCB1/SMARCA4 loss | ATRT |
| H3 K27 and G34 mutations | Small cell Glioblastoma |
| RELA fusion (L1CAM) | Supratentorial Ependymoma |
| IDH2 and IDH2 mutations | Diffuse Astrocytoma |
| ATRX/TP53 and 1p19q codeletion | Grade 2 Oligodendroglioma |
Showing case reports of paediatric ETMRs in the last 10 years [1-8].
| Sr. No. | Authors name | Year of publication | Age | Sex | Symptoms | Location | Resection | Special stains | Chemotherapy | Prognosis |
|---|
| 1 | Pfister S et al., [3] | 2009 | 2y | M | Personality changes and ataxia + ICP headache | Cerebellar Vermis | Subtotal | NeuN, Syn, INI1 positive MIB1 47% | Yes | >17 months |
| 2 | Woehrer A et al., [6] | 2011 | 2y9m | M | Increased head circumference | Left parieto-occipital lobe | Near total | INI1, Syn, NF positive | Yes | 10 months |
| 3 | Wang J et al., [2] | 2016 | 2y5m | M | Progressive visual loss | Bilateral parietal lobes | Subtotal | INI1, NeuN, NF, p53, Syn, Vim positive MIB1 70% | No | NA |
| 4 | Nobusawa S et al [4] | 2012 | 4y | M | Left hemiplegia with ICP headache | Right mid pons | Subtotal | INI1, Syn, LIN28A positive | No | NA |
| 5 | Wesseling P et al., [5] | 2014 | 2y | F | Seizures, left hemiparesis with ICP headache | Right Parieto-Occipital Lobe | Total | Syn, Vim positive | Yes | >6 months |
| 6 | Sato H et al., [8] | 2015 | 2y | M | Dysarthria, dysphagia and ataxia | Basilar Pons | Subtotal | INI1, Syn positive | Yes | 7 months |
| 7 | Tariq MU et al., [7] | 2017 | 2y9m | M | Vomiting, Gait disturbances | Intramedullary mass | Subtotal | INI1, LIN28A, NeuN, NF, Syn positive MIB1 70% | Yes | 6 months |
| 8 | Bouali S et al., [1] | 2018 | 8m | F | Left eye ptosis with ICP headache | Left cerebellar hemisphere | Subtotal | INI1, p53, NF, Syn, Vim, positive MIB1 80% | No | 1 week |
| 9 | Ganapathy S and Godhani N (Present report) | 2020 | 2y | F | Seizures | Left frontal polar lesion | Total | Syn, NeuN, INI1, Lin28A, positive, MIB1>80% | Yes | NA |
ICP: Intracranial pressure; NeuN: Neuronal nuclei; Syn: Synaptophysin; INI1: Integrase interacter-1; MIB1: Mind bomb E3 ubiquitin protein ligase 1; NF: Neurofibromatosis; LIN28A: Lin28 analog A; Vim: Vimectin
The child was reviewed and her relatives were counselled about the negative turn of events. They were advised to go for adjuvant therapy but were lost to follow-up.
Discussion
ETMR are a new classification of tumours presented in the 2016 WHO classification of Central Nervous System (CNS) tumours [9,10] The lesions are a C19-myc altered grade 4 lesions across the CNS. These include the other older lesions such as Embryonal Tumours with Abundant Neutropil and True Rosettes (ETANTR), ependymoblastomas, Medulloepitheliomas, and other c19-myc altered lesions of the CNS [10]. This diagnosis allows a significant departure from previous tumour classifications where Primitive Neuroectodermal Tumours (PNET) were combined with medulloepitheliomas and ependymoblastomas where there was no C19-myc alteration [11]. C19-myc alteration at 19q13.42 locus was observed in 93% of the tumours morphologically classified as ependymoblastomas or ETANTRs [12,13].
ETMRs are aggressive tumours [14]. They therefore, exhibit some characteristic features that are identifiable on MRI and CT scan of the brain [15]. Hence, a clear diagnosis is usually made on radiology itself before subjecting the operative specimen to histopathological and immunohistochemical analysis. We present a rare case of what radiologically appeared to be a low-grade glioma, but histologically was seen to be an aggressive ETMR. We present a radiological review of ETMR presentations and discuss its impact on treatment and prognosis.
Embryonal tumours were classified differently in the past. Different and diverse tumours such as supratentorial Primitive Neuroectodermal tumor (PNET), Medulloblastomas, Medulloepitheliomas, Neuroblastoma, Ependymoblastoma and ATRT were included together [14]. The 2016 WHO classification of tumours proposed a new classification which integrated the similarities of these lesions in a functional and simple manner [14]. If the ETMR diagnosis (C19MC alteration) is to be made according to this new classification, then 19q13.42 amplification should be assessed preferably using the Fluorescence In Situ Hybridisation (FISH). If no such Immunohistochemistry (IHC) assays are available and the diagnosis has to be made by histopathology alone then, the diagnosis of ETMR- Not Otherwise Specified (NOS) should be added to made this fact clear [15].
ETMRs are seen across the entire CNS. The most common sites include the frontal and parietal lobes as seen here in present case [14,15]. They can also be seen in the cerebellum, brainstem and even spinal cord [15]. In radiological features, the CT image shows a hyperattenuating mass in the cerebral hemisphere. MRI is generally suggestive of an aggressive lesion well-demarcated and enhancing with contrast along with significant surrounding oedema and mass effect. The lesion maybe variegated with cystic components as well. In index case, MRI of the brain showed well-defined lesions with minimal perilesional oedema with subtle enhancement on contrast not different from other similar lesion of the CNS [14], but the aggressive nature of the disease is well documented and can be discerned by indirect features such as surrounding brain oedema, contrast enhancement and rapid rate of growth. This was also not present, here in index case.
What made present case different was: 1) The entity radiologically resembled a low-grade glioma in an atypical location; 2) The lesion was slow-growing without any presenting features of raised intracranial tension or progressive neurological deficits and finally the surgical experience was also that of a low-grade lesion with poor vascularity and well-demarcated margins with a benign effect on the surrounding brain. It was only by histopathology and immunohistochemistry that the true nature of the disease was ascertained. Yet postoperative, the child recovered well and proceeded to improve and live symptom free for the next three years without incident, which is extremely rare in ETMRs.
Another complexity was what adjuvant therapy to offer this young child? Radiation therapy? Proton beam therapy? Chemotherapy? Combinations? As per literature, surgery is the primary modality, and a complete excision is of great benefit [14,15]. The most acceptable complementary therapy is chemotherapy where regimens using High Dose Chemotherapy (HDCT), Carboplatin, Cyclophosphamide and even Liposomal doxorubicin, Topotecan can be used with varied efficacy [14,15]. The average survival with chemotherapy is at most 8-12 months. Newer regimens used include chemotherapy with bone marrow transplant and radiation therapy/ Juhnke BO et al., has the series with the maximum number of patients i.e., 29, done over five years in Bonn Germany. The new protocols have a four years survival of approximately 41% which is compatible with present case report as well. Refractory progression despite treatment is known however, in 11% of patients [15].
Finally, we’d like to make a mention of proton beam radiation therapy. This is now recommended for ETMRs. IMPT is preferable (Intensity Modulated Proton Therapy) and superior to conventional radiotherapy because, it ensures better penetration, less radiation to surrounding tissue with a superior Bragg peak, easier to direct and focus, making it ideal for childhood solid brain tumours of malignant behaviour [15].
Conclusion(s)
ETMR have poor prognosis especially in children. The tumours present with features of raised intracranial tension as well as focal deficits. The radiological features are similar to other brain tumours. Thus, the final integrated diagnosis should be based on histopathology (ETMR), immunohistochemistry (LIN28A), as well as genetics (amplification of C19MC locus at 19q13.42 by FISH wherever possible) to definitively diagnose this novel aggressive brain tumour. Treatment is by surgery and chemoradiation. Proton beam radiotherapy offers some hope in this terrible disease.
ICP: Intracranial pressure; NeuN: Neuronal nuclei; Syn: Synaptophysin; INI1: Integrase interacter-1; MIB1: Mind bomb E3 ubiquitin protein ligase 1; NF: Neurofibromatosis; LIN28A: Lin28 analog A; Vim: Vimectin
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: May 14, 2020
Manual Googling: Aug 12, 2020
iThenticate Software: Sep 17, 2020 (5%)
[1]. Bouali S, Zehani A, Mahmoud M, Said IB, Kallel J, Jemel H, Embryonal tumour with multilayered rosettes: Illustrative case and review of the literatureChild’s Nervous System 2018 34(12):2361-69.10.1007/s00381-018-3972-x30215121 [Google Scholar] [CrossRef] [PubMed]
[2]. Wang J, Liu Z, Fang J, Atypical teratoid/rhabdoid tumours with multi-layered rosettes in the pineal regionBrain Tumour Pathol 2016 33:261-66.10.1007/s10014-016-0267-327307151 [Google Scholar] [CrossRef] [PubMed]
[3]. Pfister S, Remke M, Castoldi M, Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a paediatric embryonal tumour with abundant neuropil and true rosettesActa Neuropathol 2009 117:457-64.10.1007/s00401-008-0467-y19057917 [Google Scholar] [CrossRef] [PubMed]
[4]. Nobusawa S, Yokoo H, Hirato J, Analysis of chromosome 19q13.42 amplification in embryonal brain tumours with ependymoblastic multilayered rosettesBrain Pathol 2012 22:689-97.10.1111/j.1750-3639.2012.00574.x22324795 [Google Scholar] [CrossRef] [PubMed]
[5]. Wesseling P, Embryonal tumour with multilayered rosettes (ETMR): Signed, sealed, delivered Acta Neuropathologica 2014 128(2):305-08.10.1007/s00401-014-1320-025012402 [Google Scholar] [CrossRef] [PubMed]
[6]. Woehrer A, Slavc I, Peyrl A, Czech T, Dorfer C, Prayer D, Embryonal tumour with abundant neuropil and true rosettes (ETANTR) with loss of morphological but retained genetic key features during progressionActa Neuropathologica 2011 122(6):787-90.10.1007/s00401-011-0903-222057788 [Google Scholar] [CrossRef] [PubMed]
[7]. Tariq MU, Ahmad Z, Minhas MK, Memon A, Mushtaq N, Hawkins C, Embryonal tumour with multilayered rosettes, C19MC-altered: Report of an extremely rare malignant pediatric central nervous system neoplasmSAGE Open Medical Case Reports 2017 5:2050313X1774520810.1177/2050313X1774520829230288 [Google Scholar] [CrossRef] [PubMed]
[8]. Sato H, Terakawa Y, Tsuyuguchi N, Kuwae Y, Ohsawa M, Ohata K, Embryonal tumour with abundant neuropil and true rosettes in the brainstem: Case reportJournal of Neurosurgery: Pediatrics 2015 16(3):291-95.10.3171/2015.3.PEDS1472726090549 [Google Scholar] [CrossRef] [PubMed]
[9]. Louis DN, Ohgaki H, Wiestler OD, WHO classification of tumours of the central nervous system (Revised 4th edition) 2016 LyonIARC [Google Scholar]
[10]. Korshunov A, Remke M, Gessi M, Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumours with ependymoblastic rosettesActa Neuropathol 2010 120:253-60.10.1007/s00401-010-0688-820407781 [Google Scholar] [CrossRef] [PubMed]
[11]. Korshunov A, Ryzhova M, Jones DT, LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumour with multilayered rosettes (ETMR)Acta Neuropathol 2012 124:875-81.10.1007/s00401-012-1068-323161096 [Google Scholar] [CrossRef] [PubMed]
[12]. Spence T, Sin-Chan P, Picard D, CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entityActa Neuropathol 2014 128:291-303.10.1007/s00401-014-1291-124839957 [Google Scholar] [CrossRef] [PubMed]
[13]. Scheithauer BW, Development of the WHO classification of tumours of the central nervous system: A historical perspectiveBrain Pathol 2009 19:551-64.10.1111/j.1750-3639.2008.00192.x18771526 [Google Scholar] [CrossRef] [PubMed]
[14]. Pei YC, Huang GH, Yao XH, Embryonal tumour with multilayered rosettes, C19MC-altered (ETMR): A newly defined paediatric brain tumourInt J Clin Exp Pathol 2019 12(8):3156-63. [Google Scholar]
[15]. Juhnke BO, Gessi M, Mynarek M, Pietsch T, Rutkowski S, von Hoff K, PNR-25ETMR-frequency, clinical presentation, treatment and outcome of 29 patients treated within the hit trialsNeuro-Oncology 2016 18(Suppl 3):iii1110.1093/neuonc/now067.21PMC4903225 [Google Scholar] [CrossRef] [PubMed]