Rare Dysplastic Gangliocytoma of Cerebellum: A Case Report
Nidhish Kumar1, Sharvani Singh2, Bipin Kumar3
1 Senior Resident, Department of Pathology, Indira Gandhi Institute of Medical Sciences, Patna, Bihar, India.
2 Consultant Pathologist, Department of Pathology, Prakash Pathology and Radiology Pvt. Ltd., Varanasi, Uttar Pradesh, India.
3 Professor and Head, Department of Pathology, Indira Gandhi Institute of Medical Sciences, Patna, Bihar, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Nidhish Kumar, C/O Dr. Mohan Singh, Jagat Narayan Road, Kadam Kuan, Opposite Indralok Appartment, Patna, Bihar, India.
E-mail: drnidhishkumar@gmail.com
Lhermitte and Duclos first described Dysplastic Gangliocytoma in 1920. Lhermitte Duclos Disease (LDD) is an extremely rare disorder of uncertain prognosis and pathogenesis. LDD is recognised as a part of Cowden Disease, which being an autosomal-dominant phacomatosis and cancer syndrome. Till date only about 225 cases of LDD have currently been reported in medical literature. It is most commonly seen in young adults with a peak incidence in third or fourth decade with signs and symptoms of cerebellar dysfunction or increased intracranial pressure leading to obstructive hydrocephalus. A 36-year-old female came to Neuromedicine Outpatient Department (OPD) in a Tertiary Care Superspecialty Hospital with chief complaints of headache and vomiting, difficulty in swallowing liquid food since two months with no significant family history. The patient’s general condition was not good with significant weakness on presentation. The patient was conscious, oriented with pulse rate of 78/bpm, blood pressure of 118/80 mm of Hg with bilateral clear chest with normal S1 S2 sound. The Glasgow Coma Scale (GCS) was found to be Eye response-4, Verbal response-5, Motor response-6. Magnetic Resonance Imaging (MRI) of patient showed large heterogeneous non-enhancing lesion involving left cerebellar hemisphere, vermis and cerebellar peduncle with widened cerebellar folia with a “Tigroid appearance”. A diagnosis of Cerebellar Gangliocytoma was made and was treated successfully with surgery and diagnostically proven with biopsy and immunohistochemistry. The background history of Cowden syndrome was not present in index case.
Cerebellar gangliocytoma, Cowden syndrome, Obstructive hydrocephalus, Tigroid appearance
Case Report
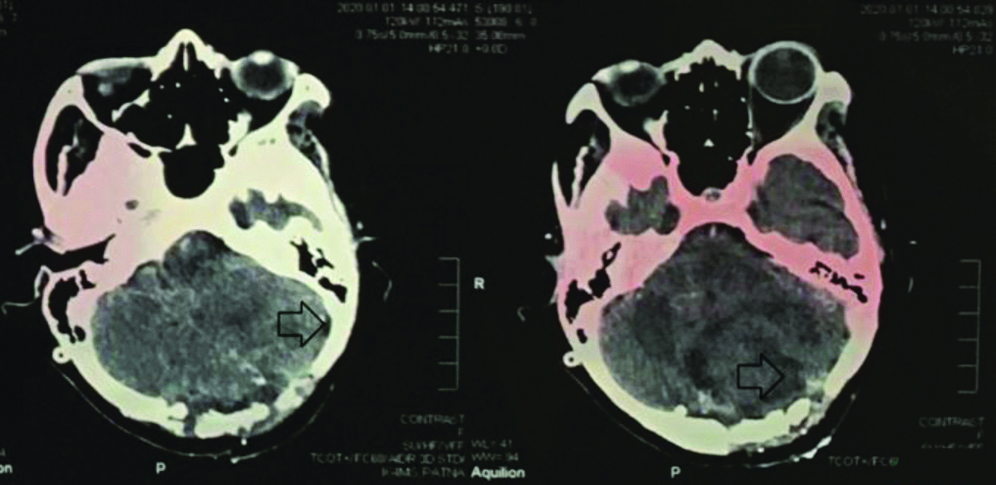
A 36-year-old female came to Neurology Medicine OPD in a Tertiary Care Superspecialty Hospital with chief complaints of headache and vomiting since two months, difficulty in swallowing liquid food since two months with no significant family history. The general condition of the patient was weak. On examination the patient was conscious, oriented with pulse rate of 78/bpm, blood pressure of 118/80 mm of Hg with bilateral clear chest with normal S1 S2 sound. The GCS was found to be E-4 V-5 M-6. The patient was referred to neurosurgery OPD and was evaluated and sent for contrast MRI in Radiology Department. A large heterogeneous non-enhancing lesion was seen involving left cerebellar hemisphere, vermis and cerebellar peduncle with widened cerebellar folia with a “Tigroid appearance”. The lesion appeared hypointense on T1W1, hyperintense on T2W1 and show hyperintensity on DW1 due to T2 shine through as shown in [Table/Fig-1,2]. The lesion showed mass effect over midbrain and fourth ventricle, causing mild dilation of bilateral lateral ventricles and third ventricle. The cerebrospinal fluid pressure was evaluated and was found to be high. A ventriculo-peritoneal shunt was placed and clear excess Cerebral Spinal Fluid (CSF) was drained out. After drainage, Non-contrast computerised tomography and Contrast-enhanced computed tomography brain was performed. It showed 5.8×5.0 cm sized mixed attenuating mass in the left cerebellar lobe which had crossed the midline. The mass had compressed fourth ventricle. Shunt was seen. Ventricles were collapsed. Bilateral cerebral hemisphere parenchymal attenuation was normal. A final impression of collapsed ventricle with shunt in situ was given. After the excess pressure was released craniotomy with gross total excision of Space Occupying Lesion (SOL) was performed after 48 hours. Preoperative haemoglobin was 8.1 gm%, Mean Corpuscular Volume (MCV) being 62.19 fL, Mean Corpuscular Haemoglobin (MCH) of 18.45 pg, Mean Corpuscular Hemoglobin Concentration (MCHC) of 29.67 g/dL, total White Blood Cell (WBC) count of 10200/cumm and platelet count of 1.79 lacs/cumm. Yellowish white cerebellar tissue was removed. Layered closure was done after a four hours surgery with a total loss of 300 mL blood. Postoperative vitals were found to be normal with GCS being E-4 V-2 M-6.
Sagittal image showing non-enhancing left cerebellar lesion (Hypo on T1 and Hyper on T2) causing compression over midbrain and fourth ventricle in the above mentioned images.
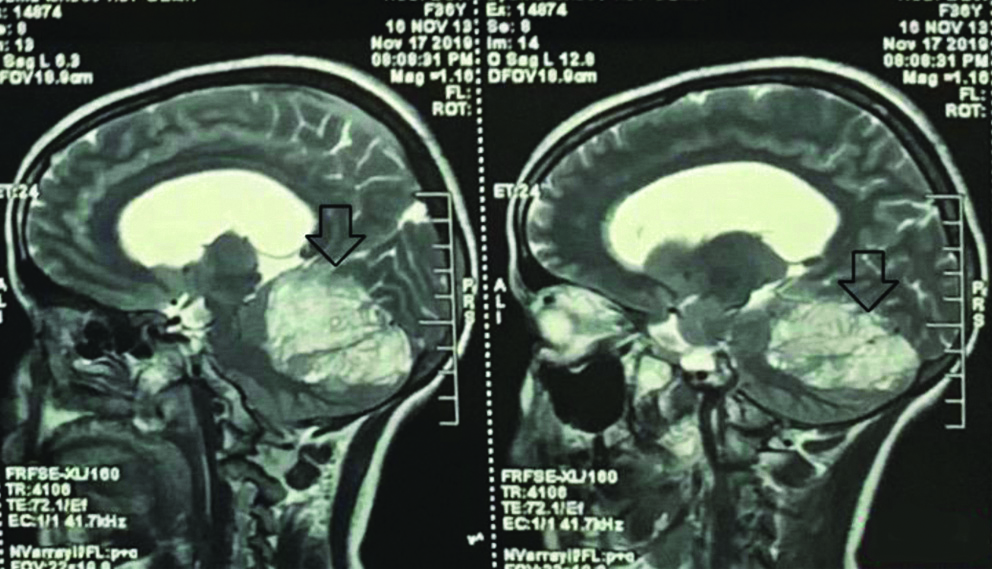
Axial image showing non-enhancing left cerebellar lesion (Hypo on T1 and Hyper on T2) causing compression over midbrain and fourth ventricle in the above mentioned images.
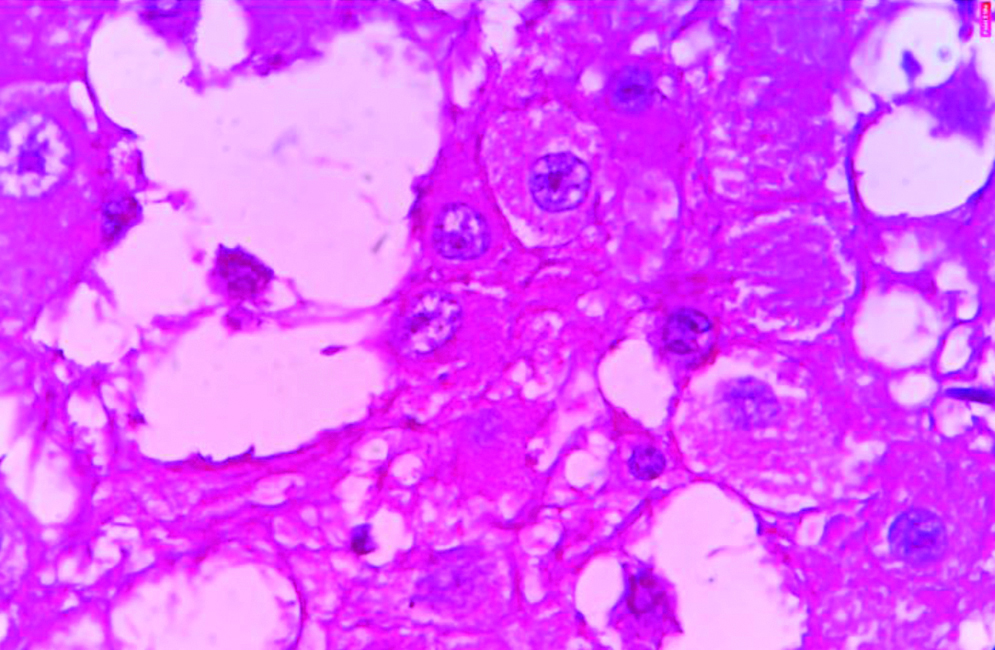
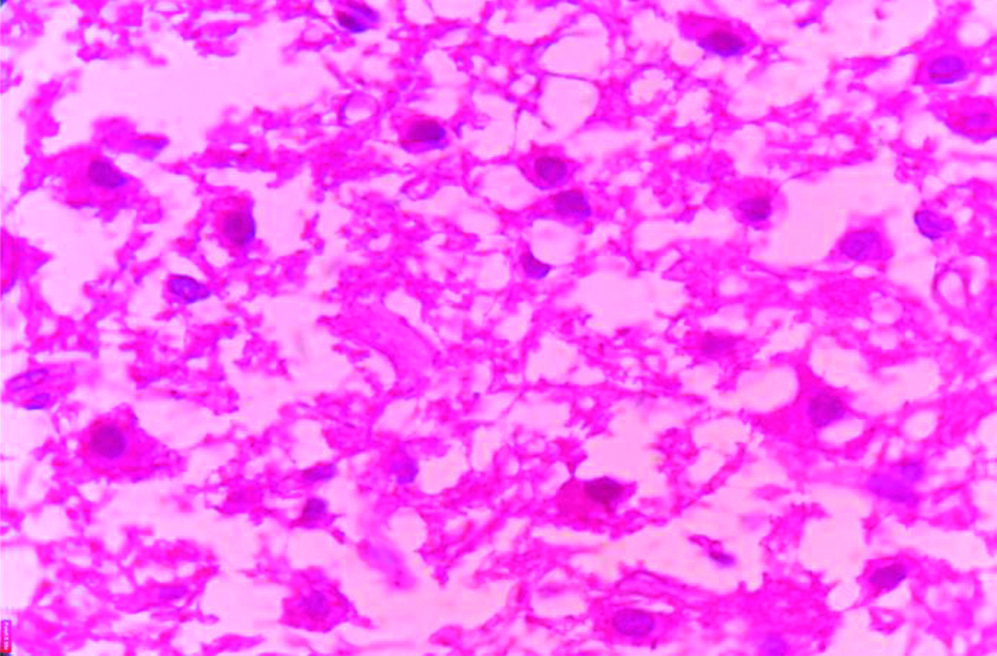
In the Pathology Department, we received a histopathological examination request of multiple pieces of cerebellar tissue altogether amounting to 3cc. Grossly, the multiple pieces were pale yellow to white with no areas of necrosis, haemorrhage and calcification. The tissue was grossed and then, processed in a benchtop tissue processor Leica TP1020 and two blocks were made. Two slides were made from the blocks and haematoxylin and eosin staining was done. The slides were made ready for microscopic examination. On microscopic examination it was seen that the cerebellar architecture was preserved focally. There was diffuse enlargement of internal granular layers and molecular layers with replacement of internal granular layers by dysplastic ganglion cells of different sizes with axonal hypermyelination of molecular layer as shown in [Table/Fig-3,4]. Clear vacuoles in white matter and molecular layer were seen. No areas of necrosis, no mitosis and no endothelial proliferation was seen. A differential diagnosis of Dysplastic Gangliocytoma and Dysplastic Cerebellar Gangliocytoma was considered. Dysplastic Gangliocytoma occurs throughout central nervous system but more than 70% of cases involve the temporal lobe. The later occurs only in cerebellum as hamartomatous cerebellar lesion characterised by enlarged cerebellar folia. Finally, the diagnosis of Dysplastic Cerebellar Gangliocytoma LCD was considered. Immunohistochemistry was performed for confirmation of dysplastic ganglion cells and the cells were found to be positive for Synaptophysin as shown in [Table/Fig-5]. Thus, the final diagnosis was made and confirmed. The stay of the patient in the hospital after surgery was uneventful and the patient was discharged after two weeks and was called for follow-up after one month. The condition of the patient was found to be stable and improving after one month of postoperative OPD visit with improvement in health, food habits and general condition of the patient. The patient is now asymptomatic after about 10 months of surgery and is doing well with improved quality of life.
X600 View of dysplastic ganglion cells showing large pleomorphic nuclei with prominent nucleoli and pink eosinophilic moderate amount of granular cytoplasm. (H&E stain).
X400 view of dysplastic ganglion cells (H&E stain).
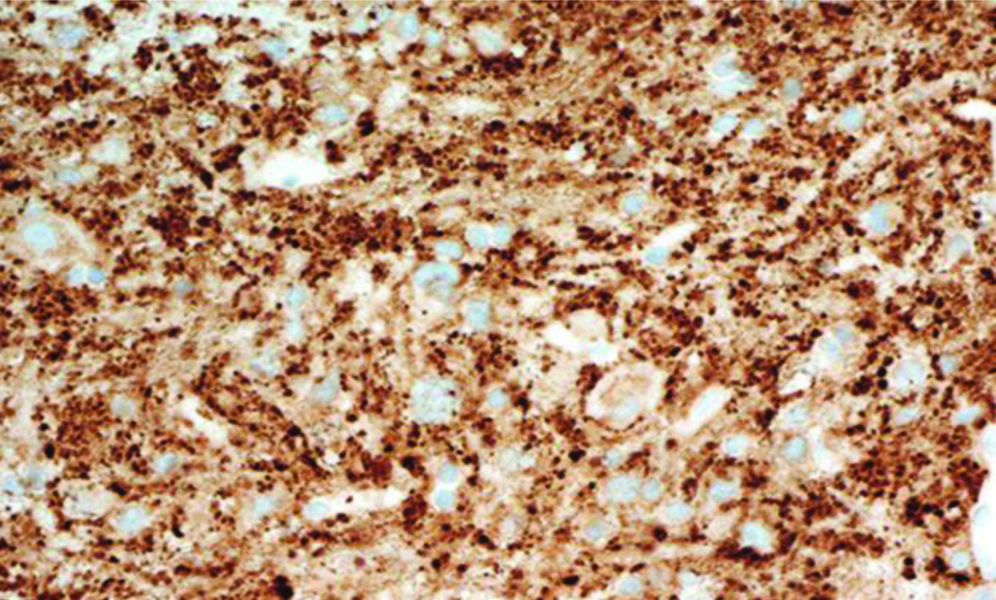
X100 view of Synaptophysin positivity in dysplastic ganglion cells showing moderate to strong, distinct cytoplasmic staining reaction (positivity).
Discussion
Lhermitte and Duclos first described Dysplastic Gangliocytoma in 1920 [1]. It is an extremely rare disorder considered as a rare cerebellar hamartoma and a part of Cowden Disease, which is an autosomal-dominant phacomatosis. LDD is sometimes called as dysplastic gangliocytoma of the cerebellum. LDD can be associated with endometrial, thyroid carcinomas and multiple hamartomas also. Mutations in the Phosphate and Tensin Homolog (PTEN) gene are associated with this “Multiple Hamartoma Neoplasia syndrome” [2,3]. Only about 225 cases of LDD have currently been reported in medical literature till date [4]. It is most commonly seen in young adults with a peak incidence in third or fourth decade of life. In patients with LDD, MRI is often diagnostic. Imaging studies have contributed in accurate diagnosis and helped in improved outcome in patients. Rare disorders such as Bannayan-Riley-Ruvalcaba and Cowden Disease are caused by mutations in the PTEN gene [5]. Most of the patients seem to be young adults presenting with signs and symptoms of cerebellar dysfunction or increased intracranial pressure leading to obstructive hydrocephalus. Patients usually present with a prolonged history which includes ill-defined neurological signs caused due to increased intracranial pressure and brainstem compression with cerebellar signs. The cerebellar signs are cranial nerve palsies with unsteadiness of gait. Cerebellar symptoms of acute onset which are characterised by rapid neurological deficit are hard to find [6].
LDD is usually associated with Cowden Syndrome [7]. Cowden Syndrome is characterised by mucocutaneous lesions, increased frequency of hamartomas and neoplasia in thyroid, breast, genitourinary organs, central nervous system and colon [7]. In present study, patient was not associated with Cowden Syndrome. Genetic mutation testing for PTEN gene should have been performed in index patient however, financial status of the patient constrained it [7]. The patient was kept close to follow-up on a yearly basis for any symptoms relating to Cowden Syndrome.
Gangliocytoma and Dysplastic Gangliocytoma of cerebellum acts as a close differential to each other and go hand in hand. Gangliocytoma being the most common type occuring throughout central nervous system and involves most commonly the temporal lobe. Dysplastic gangliocytoma of the cerebellum occurs in the cerebellum and may be associated with Cowden syndrome. Neuroimaging along with histopathological correlation with Immunohistochemistry clinches the diagnosis. In MRI, Dysplastic Gangliocytoma appears as non-enhancing cerebellar lesion (Hypo on T1 and Hyper on T2) causing compression of midbrain and fourth ventricle having classical “Tigroid Appearance” [8]. Areas of calcification and necrosis are not seen macroscopically and microscopically [8-10].
Dysplastic ganglion cells of different sizes are seen with large pleomorphic nuclei with prominent nucleoli and pink eosinophilic moderate amount of granular cytoplasm. Generally, mitosis, necrosis and neovascularity are not seen [8-10]. Immunohistochemistry shows Dysplastic Ganglion cells positive for Synaptophysin. The dysplastic ganglion cells show moderate to strong, distinct cytoplasmic staining reaction (positivity) for Synaptophysin. Clinically, patients can be asymptomatic, or they may have symptoms of ataxia, headache, cranial nerve palsy, psychic deterioration and paroxysm of vertigo [11]. In severe cases there are signs and symptoms of intracranial hypertension secondary to hydrocephalus [11]. The patients usually present with long-standing symptoms which have been present for years and indicate the slow progressive nature of the disease [11].
The background history of Cowden syndrome was not present in present case. Present case is an extremely rare case which is one of the first to be reported and documented in the eastern part of India and Nepal.
Conclusion(s)
A clinical correlation with radiological and pathological findings along with Immunohistochemistry is a must in diagnosis of LDD. Early assessment with diagnosis, surgery and close follow-up plays a very important role in the treatment, prognosis and quality of life of the patient.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Jun 15, 2020
Manual Googling: Aug 21, 2020
iThenticate Software: Sep 17, 2020 (7%)
[1]. Lhermitte J, Duclos P, Sur un ganglioneuroma diffus du cortex du cerveletBull Assoc Franc Cancer 1920 9:99-07. [Google Scholar]
[2]. Padberg GW, Schot JD, Vielvoye GJ, Bots GT, de Beer FC, Lhermitte-Duclos disease and Cowden disease: A single phakomatosisAnn Neurol 1991 29:517-23.10.1002/ana.4102905111859181 [Google Scholar] [CrossRef] [PubMed]
[3]. Moenninghoff C, Kraff O, Schlamann M, Ladd ME, Katsarava Z, Gizewski ER, Assessing a dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) with 7T MR imagingKorean J Radiol 2010 11:244-48.10.3348/kjr.2010.11.2.24420191074 [Google Scholar] [CrossRef] [PubMed]
[4]. Thomas B, Krishnamoorthy T, Radhakrishnan VV, Kesavadas C, Advanced MR imaging in Lhermitte-Duclos disease: Moving closer to pathology and pathophysiologyNeuroradiology 2007 49:733-38.10.1007/s00234-007-0241-117549467 [Google Scholar] [CrossRef] [PubMed]
[5]. Robinson S, Cohen AR, Cowden disease and Lhermitte-Duclos disease: An updateCase report and review of the literature Neurosurg Focus 2006 20(1):E6Published 2006 Jan 1510.3171/foc.2006.20.1.716459996 [Google Scholar] [CrossRef] [PubMed]
[6]. Golden N, Tjokorda MG, Sri M, Niryana W, Herman S, Management of unusual dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) in a developing country: Case report and review of the literatureAsian J Neurosurg 2016 11:17010.4103/1793-5482.14509127057227 [Google Scholar] [CrossRef] [PubMed]
[7]. Blumenthal GM, Dennis PA, PTEN hamartoma tumor syndromesEur J Hum Genet 2008 16(11):1289-300.10.1038/ejhg.2008.16218781191 [Google Scholar] [CrossRef] [PubMed]
[8]. Gupta PK, Garg S, Thanvi S, Lhermitte Duclos disease: A rare cerebellar lesion with characteristic neuroimaging featuresInt J Res Med Sci 2016 4:2478-80.10.18203/2320-6012.ijrms20161836 [Google Scholar] [CrossRef]
[9]. Reznik M, Schoenen J, Lhermitte-Duclos diseaseActa Neuropathol 1983 59:88-94.10.1007/BF006915926837278 [Google Scholar] [CrossRef] [PubMed]
[10]. Ashley DC, Zee CS, Chandrasoma PT, Segall HD, Lhermitte-Duclos disease: CT and MR findingsJ Comput Assist Tomogr 1990 14:984-87.10.1097/00004728-199011000-000222229580 [Google Scholar] [CrossRef] [PubMed]
[11]. Giorgianni A, Pellegrino C, De Benedictis A, Mercuri A, Baruzzi F, Minotto R, Lhermitte-Duclos disease. A case reportNeuroradiol J 2013 26(6):655-60.10.1177/19714009130260060824355184 [Google Scholar] [CrossRef] [PubMed]