A Rare Case of Coexistence of Homozygous β-Thalassaemia and Glucose 6 Phosphate Dehydrogenase Deficiency
Jitendar Mohan Khunger1, Monika Gupta2, Ankur Jain3, Monica Khunger Malhotra4
1 Consultant and Associate Professor, Department of Haematology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India.
2 Associate Professor, Department of Pathology, PGIMS, Rohtak, Haryana, India.
3 Assistant Professor, Department of Haematology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India.
4 Hematology-Oncology Fellow, Division of Hematology and Oncology, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Jitendar Mohan Khunger, Department of Haematology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India.
E-mail: drjmkhunger@gmail.com
β-thalassaemia is one of the most prevalent autosomal disorders worldwide. Mutations/deletions in globin gene underlie deficiencies in Haemoglobin (Hb) production, which can interfere with oxygen delivery by Hb, resulting in thalassaemias causing anaemias with a wide range of disease severity. Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is a genetic abnormality resulting in inadequate amount of G6PD in the Red Blood Cells (RBCs). In patients with G6PD deficiency, the reduced or absent activity of the enzyme in RBCs causes premature haemolysis and symptomatic anaemia. The marked oxidative stress caused by homozygous β-thalassaemia is apparently incompatible with G6PD deficiency. Here, a rare case of six-month-old male child is described who presented with severe pallor hepato-splenomegaly and these two conditions co-existed in this patient.
Anaemia, Haemoglobin, Pallor, Red blood cells
Case Report
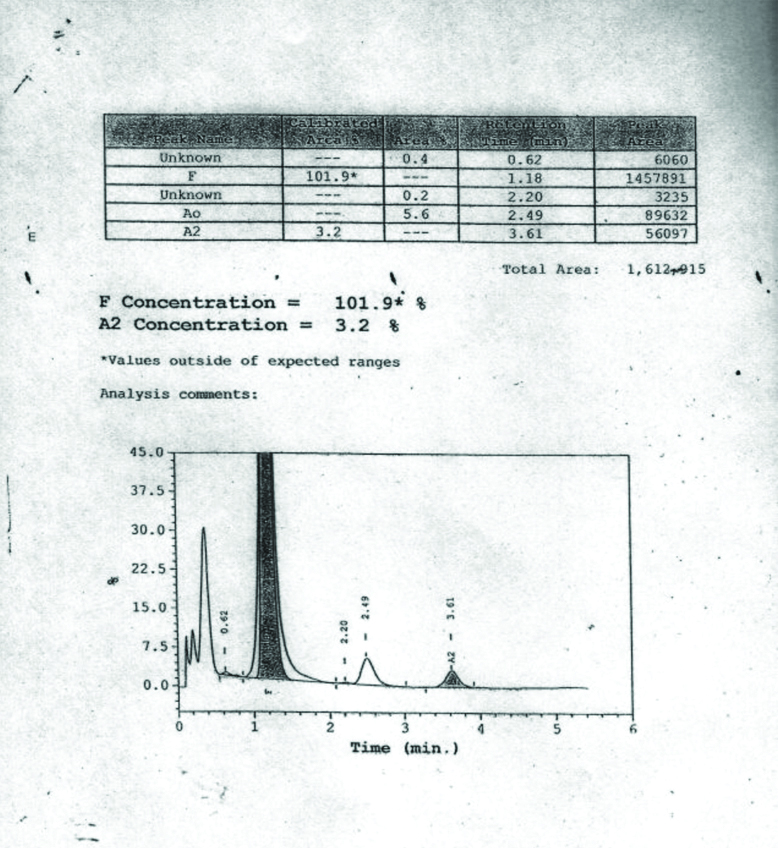
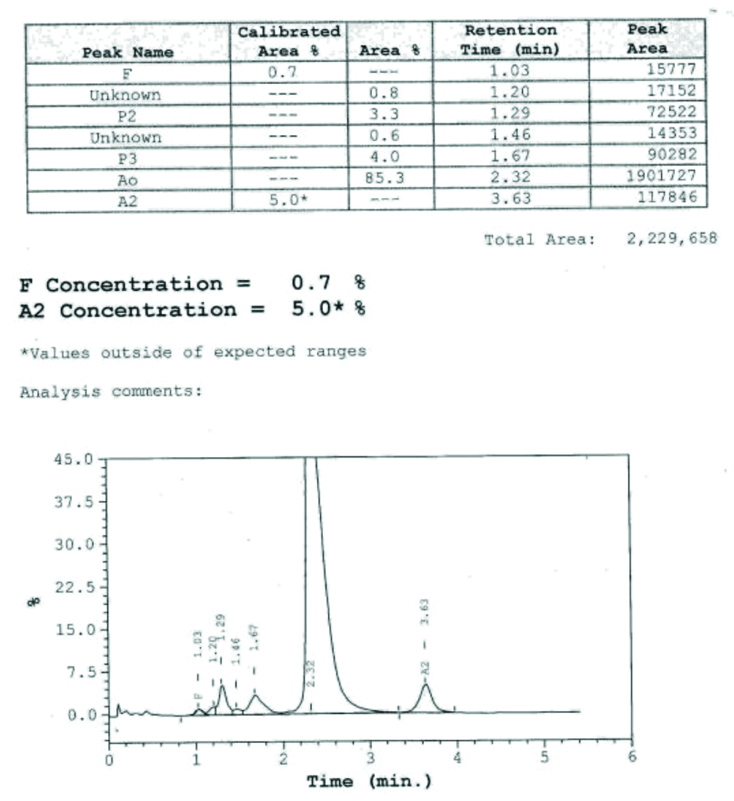
A six-month-old male child presented to the haematology outpatient department of a tertiary care centre. The parents of the child complained the child had marked pallor, not gaining weight and required frequent blood transfusions for the last one and a half months. On examination, child was having marked pallor and visible wasting with weight of 5.9 Kg. On systemic examination, there was hepato-splenomegaly. Liver was palpable 2 to 3 centimetres below the costal margin and spleen was palpable 1 centimetre below the costal margin. There was no lymphadenopathy. He had received three packed red cell transfusions in the last one and a half months. His haematologic workup showed severe anaemia with Hb of 3.8 gm/dL and RBC indices values are shown in [Table/Fig-1]. His peripheral blood smear showed severely microcytic hypochromic red cells, marked anisopoikilocytosis, polychromatic RBCs, target cells and occasional fragmented RBCs, suggestive of haemolytic picture [Table/Fig-2]. The initial workup showed that he had elevated reticulocyte count (10.8%) and elevated Lactate Dehydrogenase (LDH) levels (480 Units/L), suggestive of haemolysis. Haemoglobin analysis of patient and parents was done with High Performance Liquid Chromatography (HPLC) using equipment BioRad Variant 2. Hb HPLC of child revealed mainly HbF [Table/Fig-3] and his parental Hb HPLC study suggested heterozygous β-thalassaemia in both the parents [Table/Fig-4,5]. The provisional diagnosis of homozygous β-thalassaemia was kept in view of both parents being heterozygous β-thalassaemia. Haemolysis workup was done for the child, which included checking for Coomb’s test which was negative. G6PD enzyme activity was checked for male child, as part of his haemolysis workup. G6PD enzyme activity screening was done by methylene blue reduction test and screening for G6PD showed this enzyme deficient in RBCs of the child as shown in [Table/Fig-6]. G6PD screening result was normal in his father and his mother had G6PD deficiency in RBCs. Results of the patient and his parents are listed in the [Table/Fig-1]. After complete haemolytic work up, the child was finally diagnosed as a case of Homozygous β-thalassaemia with G6PD deficiency and managed with packed red cell transfusion. The child followed-up for packed red cells transfusion after two weeks and then lost to follow-up.
Showing results of haematological investigations of patient and parents.
| Parameter | Patient | Father | Mother |
|---|
| Sex/age | M/6 months | M/31 y | F/29 y |
| Hb (g/dL) | 3.8 | 13.0 | 10.6 |
| Hct (%) | 14.1 | 45.8 | 38.3 |
| MCV (fL) | 75.0 | 67.7 | 64.3 |
| MCH (pg) | 20.2 | 19.2 | 17.8 |
| MCHC (g/dL) | 27.0 | 28.4 | 27.7 |
| Hb typing |
| Hb A0 % | 0 | 81.6 | 85.3 |
| Hb A2 % | 3.2 | 6.4 | 5.0 |
| Hb F % | 101.9 | 1.8 | 0.7 |
| Glucose 6 phosphate dehydrogenase screening | Deficient | Normal | Deficient |
Hb-Haemoglobin; Hct-Haematocrit; MCV-Mean corpuscular volume; MCH-mean corpuscular haemoglobin; MCHC- Mean corpuscular haemoglobin concentration
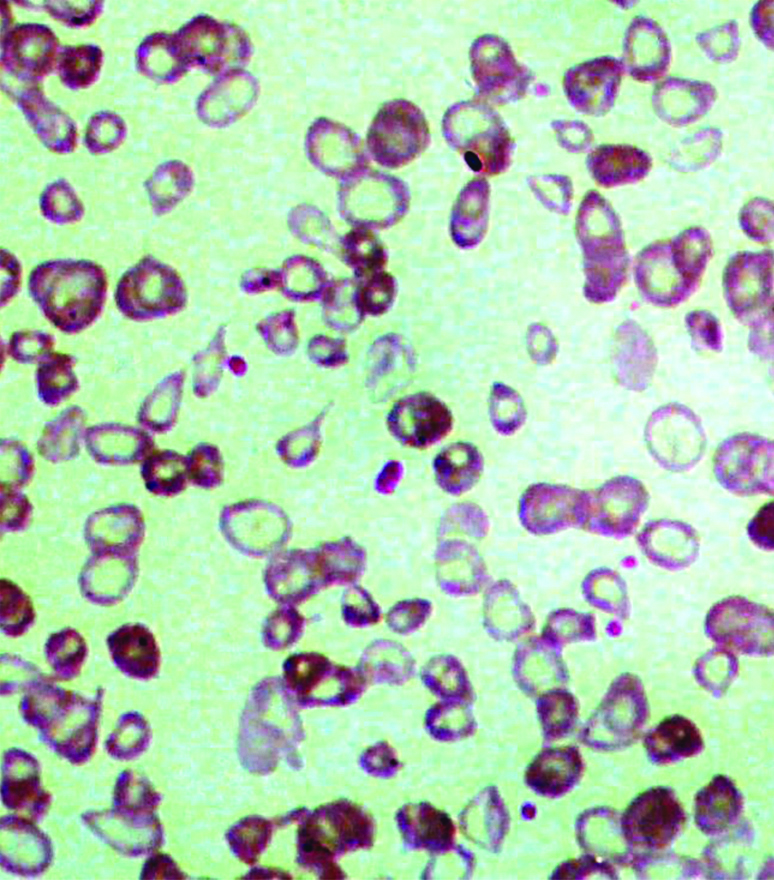
Peripheral blood smear examination of patient showing haemolytic picture (Leishman stain 10X40).
Showing Hb HPLC of patient.
Showing Hb HPLC of mother of patient.
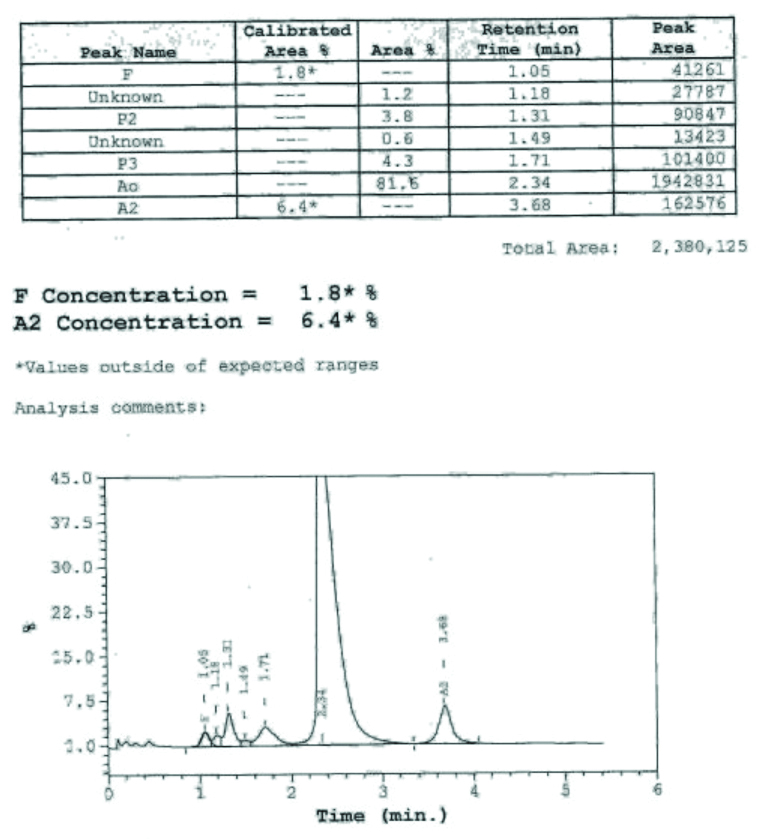
Showing Hb HPLC of father of patient.
Showing G6PD Screening test of patient.

Discussion
β-thalassaemia is an autosomal recessive disorder caused by an absence or reduction of β-globin chain synthesis. There are 270 million carriers of abnormal Hb and thalassaemias worldwide and out of these 80 millions are carriers of β-thalassaemia [1]. As per the IthaGenes database, more than 350 β-thalassaemia mutations have been reported so far [2]. The thalassaemia syndromes are named according to the globin chain affected or the abnormal haemoglobin produced. Thus, β-globin gene deletions/mutations give rise to β-thalassaemia and α-globin deletions/mutations cause α-thalassaemia. Phenotypically thalassaemia major refers to disease requiring more than eight RBC transfusions per year, and thalassaemia intermedia to disease that requires no or infrequent transfusions. Thalassaemia trait refers to carriers of this disease, such individuals have microcytosis and hypochromia but no or only mild anaemia [3-5]. The mutations/deletions result in decreased or absent production of one globin chain (α or β) and a relative excess of the other. The resulting imbalance leads to unpaired globin chains, which precipitate and cause apoptosis of red cell precursors within the marrow, termed as ineffective erythropoiesis. Of the damaged but viable RBCs that are released from the bone marrow, many are removed by the spleen or haemolysed directly in the circulation due to haemoglobin precipitants. Combined erythrocyte destruction in the bone marrow, spleen and periphery causes anaemia and ultimately, resulting in the clinical syndrome of severe thalassaemia [4,5].
Anaemia in the severe thalassaemia phenotypes necessitates multiple red cell transfusions and over time, without proper chelation, results in transfusion-associated iron overload. In addition, ineffective erythropoiesis enhances gastrointestinal iron absorption, and can result in iron overload, even in untransfused patients who have thalassaemia intermedia [6]. G6PD deficiency causes a wide range of diseases including neonatal hyperbilirubinemia, acute and/or chronic haemolysis. Disease is more severe in homozygous state and symptoms can be seen in both homozygous as well as heterozygous state [7]. G6PD catalyses the first rate limiting step in the pentose phosphate pathway that forms part of glycolysis. In the erythrocytes, the conversion of glucose-6-phosphate into 6-phosphogluconolactone, catalysed by G6PD is a very important source for the production of reduced Nicotinamide Adenine Nucleotide Phosphate (NADPH). This reduced NADPH acts by reducing glutathione and stabilising catalase. This in turn results in development of anti-oxidant properties in the RBCs. Thus, G6PD activity is needed for erythrocytes to withstand oxidative stress [7]. Deficiency of G6PD enzyme activity impairs the erythrocyte’s ability to remove deleterious oxygen species, leading to premature lysis and cell death [7]. Magnani M et al., showed that in patients who were heterozygous for β-thalassaemia and had simultaneous G6PD deficiency, the RBC contained practically undetectable levels of NADPH [8]. Many studies done in western cohort reported the co-inheritance of β-thalassaemia trait with G6PD deficiency. The study done by Troitskaia OV et al., found that the β-thalassaemia trait and G6PD deficiency resulted in a severe clinical manifestation in children as compared to adults [9]. Similar findings were seen in the study done by Giles E et al., in New guinea [10].
Hematological parameters between thalassaemia carriers with G6PD deficiency and those without G6PD deficiency were compared in studies published in Hb journal in year 2013 [11] and 2017 [12]. Similarly, G6PD deficiency co-inherited with Hb E heterozygotes, β-thalassaemia trait, and β-thalassaemia/Hb E, α-thalassaemia-2 trait, and Hb H disease studied in year 2018 [13], for basic haematologic parameters.
It was found that Hb, haematocrit, mean cell volume, and mean cell Hb of G6PD deficiency co-inherited with asymptomatic thalassaemia carriers, showed significantly lower mean values, compared to, carriers with only the same thalassaemia genotypes without G6PD deficiency [13]. It was concluded in their study that, G6PD deficiency co-inherited with thalassaemia carriers in males was present in 10% of the participants, resulting in worsening of RBC pathology compared with inheritance of thalassaemia trait alone [13]. There is paucity of literature regarding the co-inheritance of homozygous β-thalassaemia along with G6PD deficiency. However, it is well known that sickle cell trait and G6PD deficiency may be present in malaria endemic areas [14]. It is also known from genetic sequencing study done in the cord blood samples in Riyadh [15], that HbS and G6PD deficiency may co-exist.
Conclusion(s)
The authors conclude by saying that the combination of β-thalassaemia major and G6PD deficiency is a very rare co-inheritance that can be confused with various disorders inherited congenitally like congenital dyserythropoeitic anaemia and diamond blackfan anaemia. Thus, this case report emphasises the fact that the disease may be kept as an important differential possibility when considering similar clinical presentation, encountered in this age group.
Hb-Haemoglobin; Hct-Haematocrit; MCV-Mean corpuscular volume; MCH-mean corpuscular haemoglobin; MCHC- Mean corpuscular haemoglobin concentration
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. NA
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: May 18, 2020
Manual Googling: Jul 31, 2020
iThenticate Software: Sep 16, 2020 (3%)
[1]. De Sanctis V, Kattamis C, Canatan D, Soliman AT, Elsedfy H, Karimi M, β-thalassaemia distribution in the old world: An ancient disease seen from a historical standpointMediterr J Hematol Infect Dis 2017 9(1):e201701810.4084/mjhid.2017.01828293406 [Google Scholar] [CrossRef] [PubMed]
[2]. Kountouris P, Lederer CW, Fanis P, Feleki X, Old J, Kleanthous M, IthaGenes: An interactive database for haemoglobin variations and epidemiologyPLoSOne 2014 9(7):e10302010.1371/journal.pone.010302025058394 [Google Scholar] [CrossRef] [PubMed]
[3]. Deisseroth A, Nienhuis A, Lawrence J, Giles R, Turner P, Ruddle FH, Chromosomal localization of human β-globin gene on human chromosome 11 in somatic cell hybridsProc Natl Acad Sci USA 1978 75(3):1456-60.10.1073/pnas.75.3.1456274732 [Google Scholar] [CrossRef] [PubMed]
[4]. Cunningham JM, Macklin EA, Neufeld EJ, Cohen AR, Complications of β-thalassaemia major in North AmericaBlood 2004 104(1):34-39.10.1182/blood-2003-09-316714988152 [Google Scholar] [CrossRef] [PubMed]
[5]. Cao A, Carrier screening and genetic counseling in β-thalassaemiaInt J Hematol 2002 76(suppl 2):105-13./10.1007/BF0316509812430909 [Google Scholar] [CrossRef] [PubMed]
[6]. Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, Ineffective erythropoiesis in β-thalassaemia is characterized by increased iron absorption mediated by down regulation of hepcidin and upregulation of ferroportinBlood 2007 109(11):5027-35.10.1182/blood-2006-09-04886817299088 [Google Scholar] [CrossRef] [PubMed]
[7]. Frank JE, Diagnosis and management of G6PD deficiencyAm Fam Physician 2005 72(7):1277-82. [Google Scholar]
[8]. Magnani M, Stocchi V, Canestrari F, Cucchiarini L, Stocchi O, Coppa GV, Redox and energetic state of red blood cells in G6PD deficiency, heterozygous beta-thalassaemia and the combination of bothActa Haematol 1986 75(4):211-14.10.1159/0002061273096052 [Google Scholar] [CrossRef] [PubMed]
[9]. Troitskaia OV, Ermil’chenko GV, Levitskaia SV, Varzieva LK, Beta-thalassaemia and glucose-6-phosphate dehydrogenase deficiencyKlin Lab Diagn 1996 (6):17-24. [Google Scholar]
[10]. Giles E, Curtain CC, Baumgarten A, Distribution of β-thalassaemia trait and erythrocyte glucose-6-phosphate dehydrogenase deficiency in the Markham river valley of New GuineaAm J Phys Anthropol 1967 27:83-88.10.1002/ajpa.13302701106058057 [Google Scholar] [CrossRef] [PubMed]
[11]. Pornprasert S, Phanthong S, Anaemia in patients with coinherited thalassaemia and glucose-6-phosphate dehydrogenase deficiencyHemoglobin 2013 37(6):536-43.10.3109/03630269.2013.81955823944358 [Google Scholar] [CrossRef] [PubMed]
[12]. Banyatsuppasin W, Jindadamrongwech S, Limrungsikul A, Butthep P, Prevalence of thalassaemia and glucose-6-phosphate dehydrogenase deficiency in newborns and adults at the Ramathibodi Hospital, Bangkok, ThailandHemoglobin 2017 41(4-6):260-66.10.1080/03630269.2017.140202629251006 [Google Scholar] [CrossRef] [PubMed]
[13]. Pengon J, Svasti S, Kamchonwongpaisan S, Vattanaviboon P, Hematological parameters and red blood cell morphological abnormality of Glucose-6-Phosphate dehydrogenase deficiency co-inherited with thalassaemiaHematol Oncol Stem Cell Ther 2018 11(1):18-24.10.1016/j.hemonc.2017.05.02928641093 [Google Scholar] [CrossRef] [PubMed]
[14]. Bienzle U, Sodeinde O, Effiong CE, Luzzatto L, Glucose 6-phosphate dehydrogenase deficiency and sickle cell anaemia: Frequency and features of the association in an African communityBlood 1975 46(4):591-97.10.1182/blood.V46.4.591.5911174693 [Google Scholar] [CrossRef] [PubMed]
[15]. Al-Nuaim L, Talib ZA, el-Hazmi MA, Warsy AS, Sickle cell and G6PD deficiency gene in cord blood samples: Experience at King Khalid University Hospital, RiyadhJ Trop Pediatr 1997 43(2):71-74.10.1093/tropej/43.2.71 [Google Scholar] [CrossRef]