The MBCTs forms a significant fraction of paediatric tumours, accounting for about one-third of paediatric malignant neoplasms. They are heterogeneous tumours with overlapping diagnostic features. However, most cases, although aggressive, are curable with advanced chemotherapeutic regimens that demand exact diagnosis [1]. Cytologically, most of these tumours represent more or less small-intermediated, round/oval and relatively undifferentiated cells that make definite diagnosis difficult on Haematoxylin and Eosin (H&E) stained sections. Careful handling with well sampled biopsy specimens in addition to perfectly processed and properly stained H&E tumour sections as well as clinico/radiological acumen have a great impact on the relative likelihood of MBCT diagnosis [2]. In 15-20%, there is a need for further evaluating tests. For IHC, it is important to employ panels of antibodies (rather than a single marker) and to integrate all these available evidences for a conclusive diagnosis. These antibodies include different carcinoma markers (Cytokeratin (CK), Epithelial Membrane Antigen (EMA)), lymphoma/leukaemia markers (CD45, CD20, CD3, PAX5 (Paired Box 5), Terminal Deoxynucleotidyl Transferase (TDT), CD34, CD117), sarcoma markers (Vimentin, Desmin, MyoD1, Myogenin, Smooth muscle actin), Glial Fibrillary Acidic Protein (GFAP), neuroblastoma marker (CD56, synaptophysis) and different other markers accordingly.
In Duhok, Iraq, detailed paediatric MBCTs were not dealt yet. This study was an attempt to apply traditional and update antibodies to categorise MBCTs among paediatric age groups, by integration of clinical and morphological data with immunohistochemical results in this particular locality.
Materials and Methods
The present study was a cross-sectional study done over a 11 year-period, from January 2009 to December 2019, after obtaining Ethical clearance (number 2422) enrolled 120 children, up to 15-year-old, with morphologically diagnosed MBCTs. Ethical clearance was obtained and consent was taken. Cases were received in the Department of Histopathology in Central General and Vin Private Laboratories, Duhok, Iraq. The study was conducted in Vin Private Laboratories. All samples were subjected to panels of immunomarkers depending on the tumour morphology, patient’s age and tumour location. Extra panels of antibodies were then added accordingly, [Table/Fig-1] as described previously [3-12]. The Autostainer Link Instrument, immunohistochemical staining was applied using the UltraVision Localizer Performance (LP) Large Volume Detection System and Horseradish Peroxidase (HRP) Polymer (Ready-To-Use) from Thermo Fisher Scientific® as described by the manufacturer’s recommendations. Special stains (PAS/PASD, reticulin and Trichrome) were performed in some cases to detect glycogen and fibrous tissue.
Detailed antibodies used for Malignant Blue Cell Tumour (MBCT) categorisation.
| Tumour | Immunohistochemical panel |
|---|
| Ewing’s/PNET (Primitive Neuroectodermal tumour) | Vimentin, CD99, CD56, Synaptophysin, chromogranin, NSE, FLI-1. PAS+/PASD- |
| Neuroblastoma | Synaptophysin, CD56, NSE, Chromogranin, CD99. |
| Wilms tumour | Pankeratin, WT1. |
| Rhabdomyosarcoma | Vimentin, Desmin, Myogenin, MyoD1, Myoglobin, CD99. |
| Lymphoblastic lymphoma | CD45, CD2, CD3, CD20, PAX5, CD79a, TDT, CD99, CD34, CD10. |
| Burkitt’s and DLBL | CD45, CD20, PAX5, CD79a, CD10, BCL2, BCL6, MUM1, Ki67. |
| Lymphatic leukaemia | B (CD20, PAX5, CD79a) and T (CD3, CD2, CD4, CD5, CD8) cell lineage. |
| Myeloid leukaemia | CD14, CD34, Myeloperoxidase, CD117. |
| Retinoblastoma | Synaptophysin, Leu7. |
| Glioblastoma | GFAP. |
| Ependymoma | GFAP, EMA, CD99. |
DLBL: Diffuse large B-cell lymphoma; CD: Cluster of differentiation, FLI-1: Friend leukemia integration 1 transcription factor, NSE: Neuron specific enolase, WT1: Wilm’s tumour 1, PAX: Paired box, TDT: Terminal deoxynucleotidyl transferase, BCL: B-cell lymphoma, Ki67: Proliferative index, GFAP: Glial fibrillary acidic protein and EMA: Epithelial membrane antigen
Results
In this study, 120 children, aged 1 month to 15 years (mean: 8.49 years) were enrolled included 66 males and 54 females with 1.2:1 male to female ratio. The peak tumour burden for both sexes was at 11-15 years and the least represented cases were among infancy [Table/Fig-2]. No significant age and sex differences were demonstrated (p>0.05).
Malignant Blue Cell Tumours (MBCT) distribution with age intervals and sex.
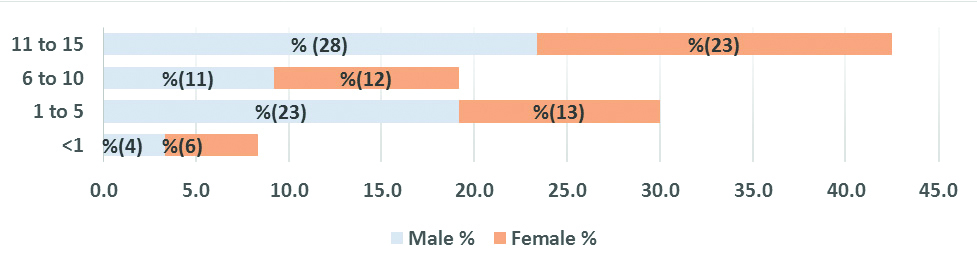
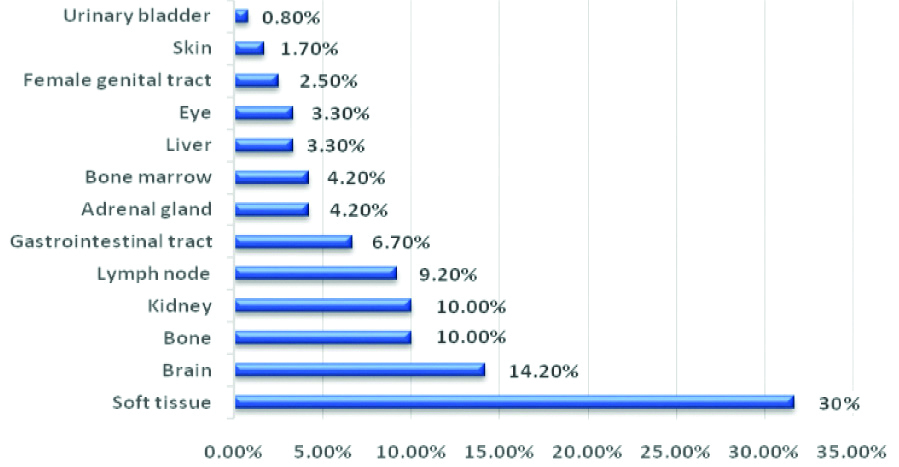
Regarding tumour location as shown in [Table/Fig-3], soft tissue was the top location (30.0%) followed by brain (14.2%). Less common tissues/sites included gastrointestinal tract (6.7%), skin (1.7%) and urinary bladder (0.8%).
Malignant Blue Cell Tumours (MBCT) distribution and location.
Malignant Blue Cell Tumour (MBCT) Types
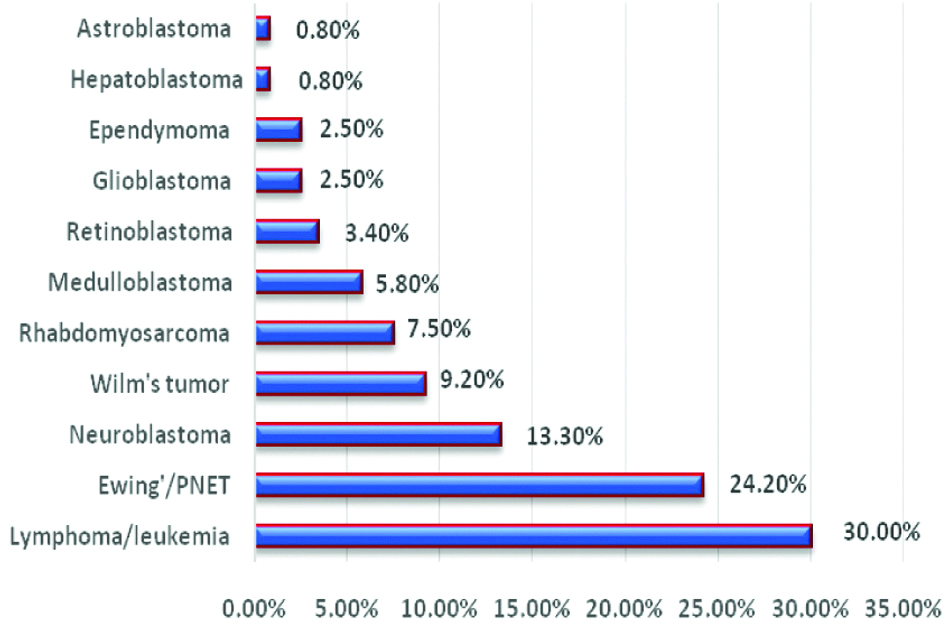
As illustrated in [Table/Fig-4], lymphoma/leukaemia formed the commonest category, followed by Ewing’s/PNET, Neuroblastoma, Wilm’s tumour, Rhabdomyosarcoma and Medulloblastoma. The remainders comprised Retinoblastoma, Glioblastoma and Ependymoma. Hepatoblastoma and Astroblastoma formed the least frequent tumours.
Percentages of Malignant Blue Cell Tumour (MBCT) types.
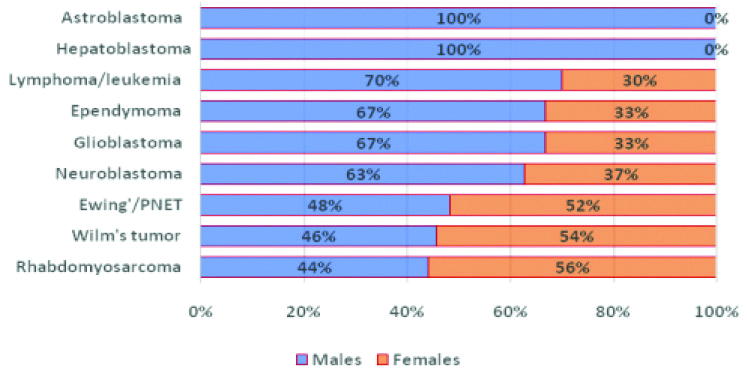
Considering sex distribution, males formed 70% of Leukaemia/lymphoma, 67% of each of Ependymoma and Glioblastoma, 63% of Neuroblastoma, 48% of Ewing’s/PNET, 46% of Wilm’s tumour and 44% of Rhabdomyosarcoma. Both Astroblastoma and Hepatoblastoma patients were males. The remainders were females [Table/Fig-5]. No significant gender differences were demonstrated.
Malignant Blue Cell Tumour (MBCT) types and sex distribution.
Lymphoma/Leukaemia
Out of 36 Lymphomas/leukaemia cases, 26 (72.2%) were B-cell Non-Hodgkin Lymphoma (NHL) and 4 (11.1%) were T-cell type NHL. The remaining 6 (16.7%) cases were Leukaemias. About 72% of Leukaemia/lymphoma were extranodal and about 28% were nodal. Most patients (70%) were males and 61% presented at 11-15 years age interval [Table/Fig-6].
Age and sex distribution of Lymphoma/leukaemia.
| Type | Sex: Number (%) | Total | Age interval (years): Number (%) |
|---|
| Male | Female | Number (%) | <1 | 1 to 5 | 6 to 10 | 11 to 15 |
|---|
| B-cell NHL | 16 (44.4) | 10 (27.8) | 26 (72.2%) | 0 (0.0) | 4 (11.1) | 7 (19.4) | 15 (41.7) |
| Leukaemia | 5 (14.0) | 1 (2.8) | 6 (16.7) | 1 (2.8) | 0 (0.0) | 1 (2.8) | 4 (11.1) |
| T-cell NHL | 4 (11.1) | 0 (0.0) | 4 (11.1) | 0 (0.0) | 1 (2.8) | 0 (0.0) | 3 (8.2) |
| Total | 25 (70) | 11 (30) | 36 (100) | 1 (2.8) | 5 (13.9) | 8 (22.3) | 22 (61) |
Histopathology and Immunohistochemical Results
Of the 26 B-cell NHL, 14 (53.8%) were DLBL including a single case with focal follicular pattern, 8 (30.8%) Burkitt’s lymphoma and 4 (15.4%) B-Lymphoblastic Lymphoma (LBL). All T-cell NHL were T-cell lymphoblastic lymphomas. The six leukaemia cases included four Acute Myeloid Leukaemia (AML) and two Chronic Myeloid Leukaemia (CML) including a case of Leukaemia cutis.
Diffuse Large B-cell Non-Hodgkin Lymphoma
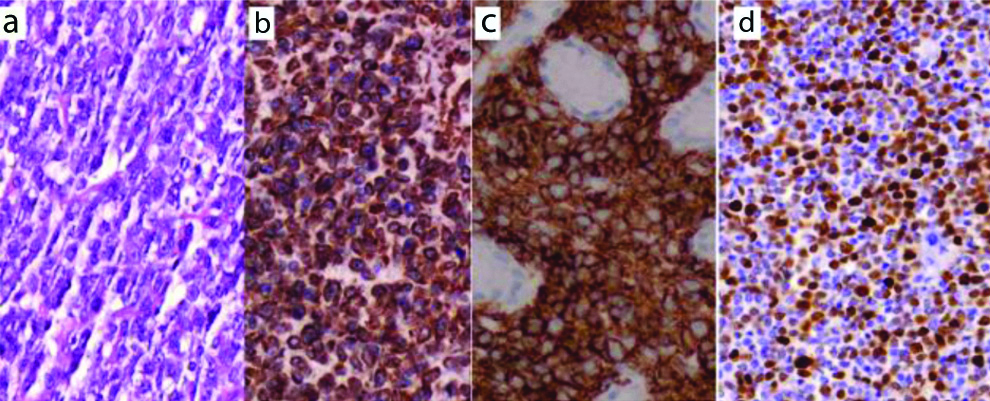
Morphologically, 14 cases (6 nodal and 8 extranodal) showed a diffuse infiltration with monotonous medium to large lymphoid cells with a frequent mitotic figure, vesicular nuclei and prominent 1-2 nucleoli. Necrosis was variable. Focally, a single nodal case exhibited follicular growth pattern. As illustrated in [Table/Fig-7], all DLBL were CD45 positive/CD3 negative. As well, they were positive for at least two of CD20, CD79a and PAX5. Individually speaking, DLBL cases were positive for CD20 (92.9%), PAX5 and CD79a (85.7% each), Bcl2 (78.6%), CD10 (57.1%), Bcl6 and MUM1 (42.9% each). Apart from one case with about 60% proliferative index (Ki67), all showed more than 90% Ki67 nuclear positivity [Table/Fig-8].
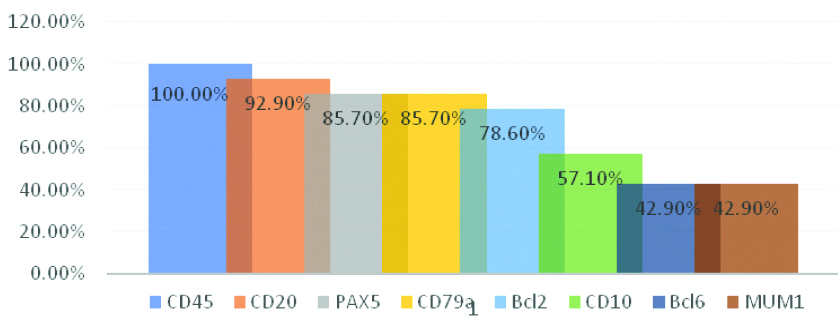
Diffuse Large B-cell lymphoma morphology, a (H&E, X400), positive for: b: CD20, c: Bcl2, d: About 60% Ki67 (b-d: IHC, X400).
Burkitt’s Lymphoma
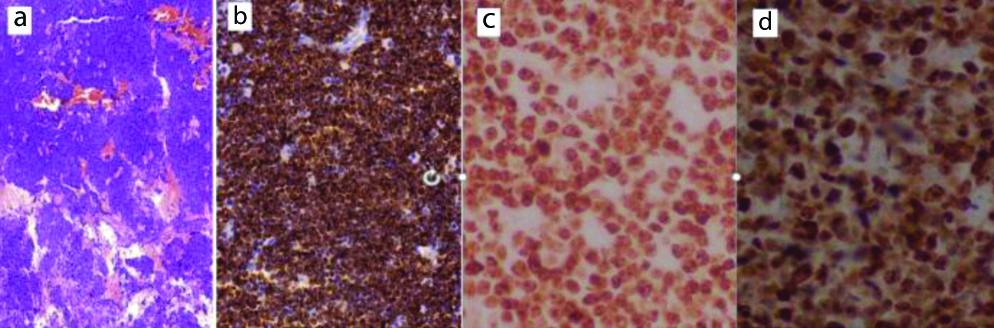
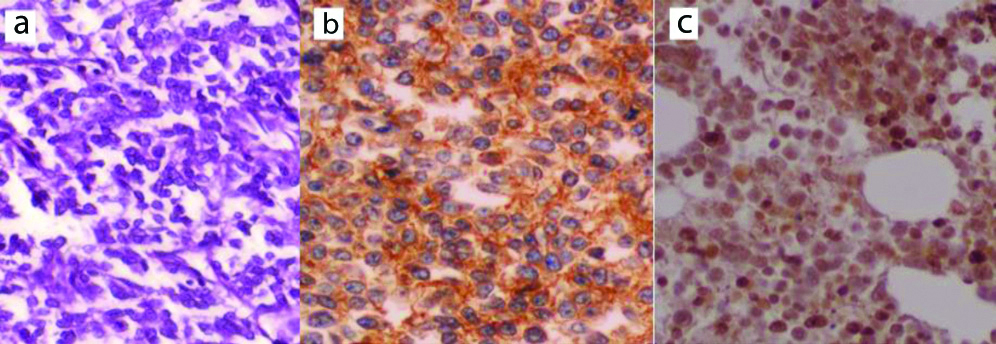
Eight cases presented a diffuse infiltration by small-medium sized lymphoid cells showing brisk mitotic activity within starry sky background. The cells showed round nuclei with multiple tiny nucleoli and scant cytoplasm. All cases were positive for CD45, CD20, PAX5, CD10 and Bcl6 [Table/Fig-9], while negative for CD3 and Bcl2. The proliferative index (Ki67) was positive in 100% of tumour cells.
Burkitt’s lymphoma morphology: a) H&E (X40), positive for; b) CD20 (IHC, X100); c) Bcl6 (IHC, X400); d) 100% Ki67 positivity (IHC, X400).
Lymphoblastic Lymphoma
As shown in [Table/Fig-10], eight cases showed a diffuse pattern infiltration with monotonous small sized lymphoid cells with scant cytoplasm and nuclei showing dusty chromatin and irregular nuclear contour. Nucleoli were inconspicuous. The background showed a starry sky appearance. Immunohistochemically, four cases were committed to B-cell immunoprofile (CD79a, CD10). PAX5 and CD20 were positive in three cases only. The other four cases represented T-cell lymphoma (CD3 positive).
Lymphoblastic lymphomas LBL categorisation.
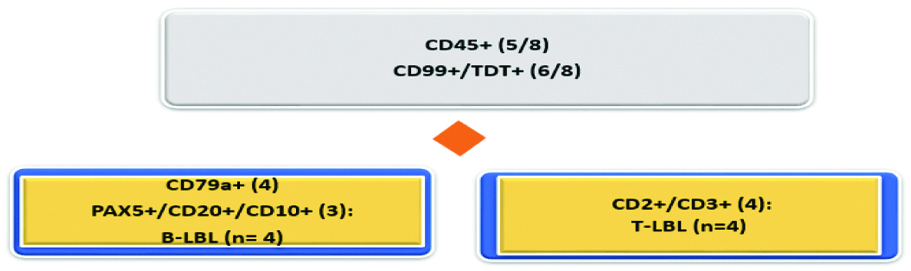
TDT and CD99 were positive in 6/8. Only 5/8 showed CD45 positivity [Table/Fig-11]. As well a single CD45 positive/Vimentin negative case also expressed nuclear Friend Leukaemia Integration-1 (FLI-1) protein.
Lymphoblastic lymphoma morphology: a) H&E (X100), positive for; b) CD99 (IHC, X400); c) CD10 (IHC, X400); d) TDT (IHC, X400).
Sarcomas
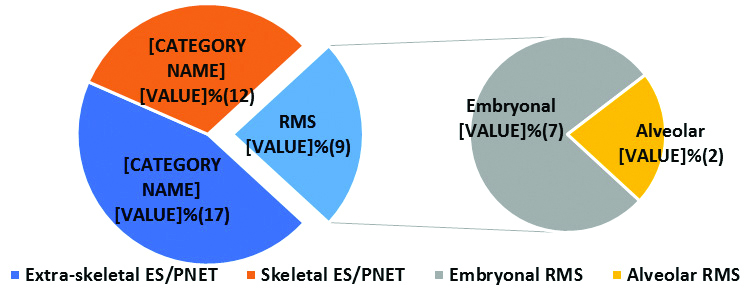
Ewing’s sarcoma/PNET formed the commonest sarcomatous tumours, 29 (24.2% of total MBCT and 76.3% out of sarcoma cases) with 44.7% extraskeletal and 31.6% skeletal, followed by Rhabdomyosarcoma 9 (7.5% of total MBCT and 23.7% out of sarcoma cases). Their percentages and locations are summarised in [Table/Fig-12].
Percentages of sarcoma subtypes.
ES: Ewing’s; RMS: Rhabdomyosarcoma
Ewing’s/PNET
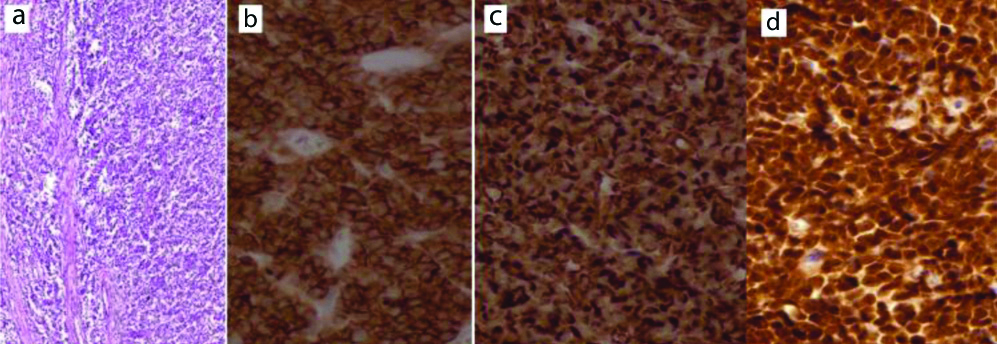
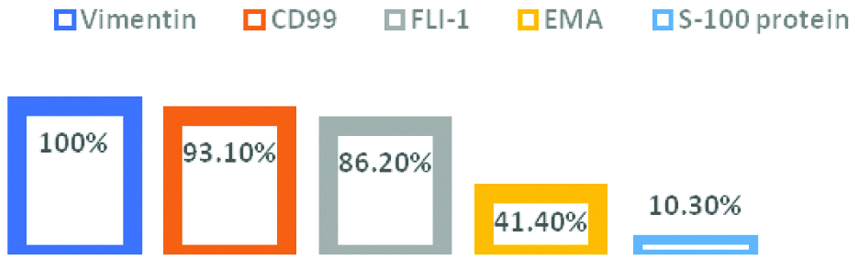
Diagnosis of Ewing’s/PNET was given for 29/120 cases presented as nests, trabeculae and solid sheets of small cells with scant cytoplasm, round to oval nuclei and distinct nucleoli with evidence of extensive necrosis leaving perivascular rims of tumour cells. Mitosis was brisk with evidence of atypical mitoses. Twenty cases were PAS positive/PASD negative referring to cytoplasmic glycogen. As shown in [Table/Fig-13], the tumour cells stained strongly for Vimentin (100%), CD99 (93.1%) and Fli-1 (86.2%). EMA and S-100 protein were positive in 41.4% and 10.3%, respectively [Table/Fig-14].
Ewing sarcoma/PNET immunoresults.
Ewing’s/PNET morphology: a) H&E (X400); b) positive for CD99; c) Fl1-1 (IHC, X400).
Neuroblastoma
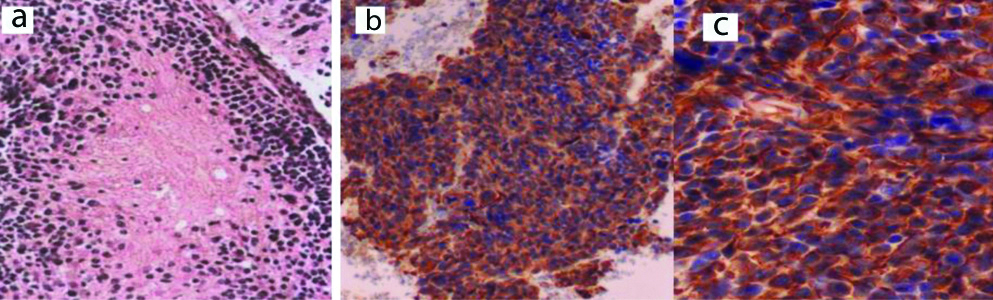
Sixteen MBCT cases grew as sheets and nests of small blue cells with variable neuropil elements with some Homer Wright pseudorosettes. Variable degrees of necrosis and mitotic activity were noticed. These cases were put under the diagnosis of neuroblastomas although six cases were demonstrated at unusual locations (lymph node, liver, bone and bone marrow), as shown in [Table/Fig-15], they stained positive for at least two of endocrine markers (Synaptophysin, NSE, CD56 and Chromogranin). After thorough clinical evaluation, associated primary tumours were discovered in the unusually located cases, and thus considered as metastatic. At time of diagnosis, two cases were associated with elevated Vanillylmandelic acid in urine.
Neuroblastoma morphology: a) H&E (X100); b) Synaptophysin; c) CD56 (IHC: B, X100; C, X400).
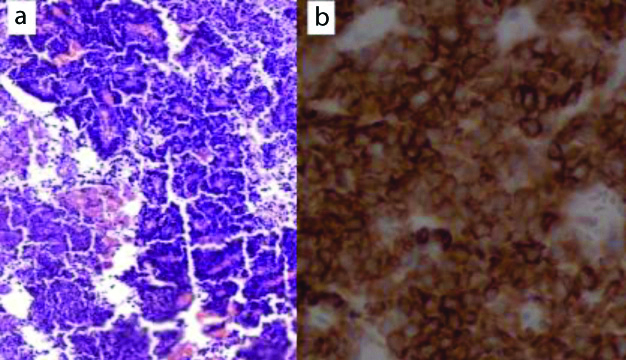
Wilm’s Tumours
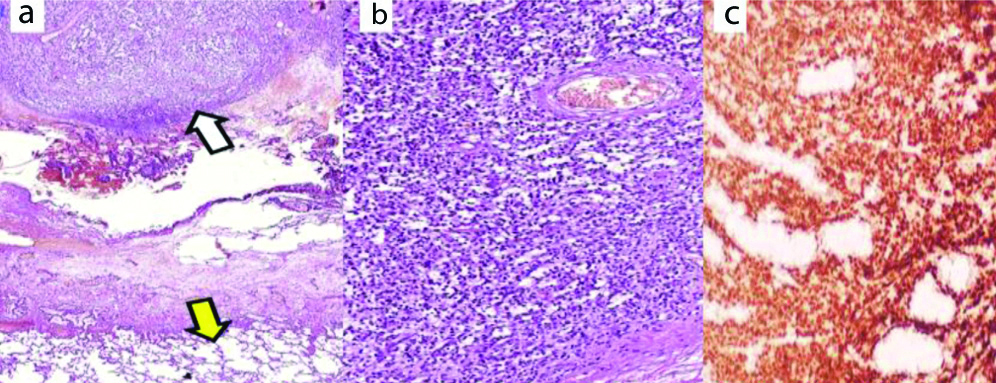
Final diagnosis of Wilm’s tumour was given for 11/120 cases forming solid sheets and diffuse infiltration by small round to oval cells with scant cytoplasm. In four cases, foci of epithelial and/or mesenchymal elements could be detected somewhere within the undifferentiated blastemal tumour cells. One case: nine-year-old boy with a recurrent Wilm’s tumour was diagnosed at a remote site (lung). It showed a strong nuclear positivity for WT1 [Table/Fig-16] and aberrantly positive for CD99, while negative for CD45, endocrine and epithelial markers.
Wilm’s tumour nodule (white arrow) metastasizing to the lung (yellow arrow): a) H&E, X40; b) H&E, X100; c) Positive for WT-1 (IHC, X100).
Rhabdomyosarcoma
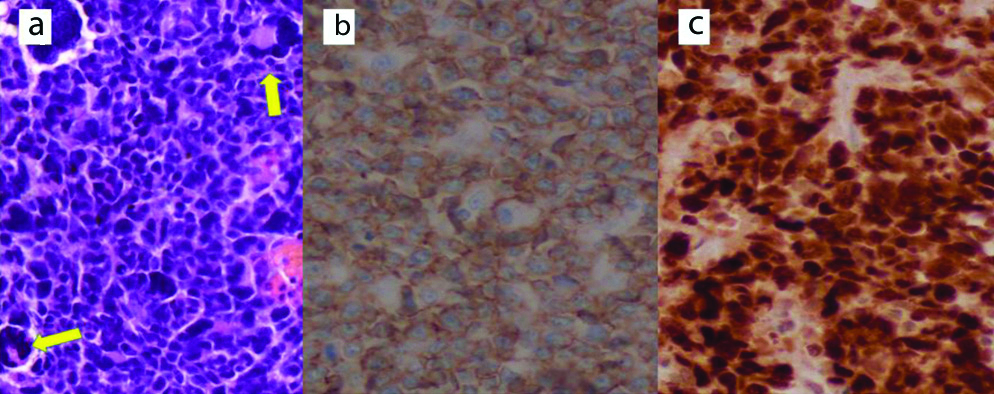
Nine out of 120 cases were classified as Rhabdomyosarcoma. Histomorphologically, seven cases presented as solid growths and singly scattered undifferentiated small round cells within a myxoid. These cases were stratified as Embryonal Rhabdomyosarcoma. In the other two cases typed as Alveolar Rhabdomyosarcoma, the malignant blue cells gave alveolar growth pattern with scattered multinucleated giant cells. Foci of necrosis were demonstrated. The cells of the nine cases expressed Vimentin, Desmin, Myogenin, MyoD1, Myoglobin and, only two cases for CD99 [Table/Fig-17].
Rhabdomyosarcoma morphology, showing scattered multinucleated giant cells (arrows): a) (H&E, X400), positive for; b) CD99; c) MyoD1 (IHC: X400).
Medulloblastoma
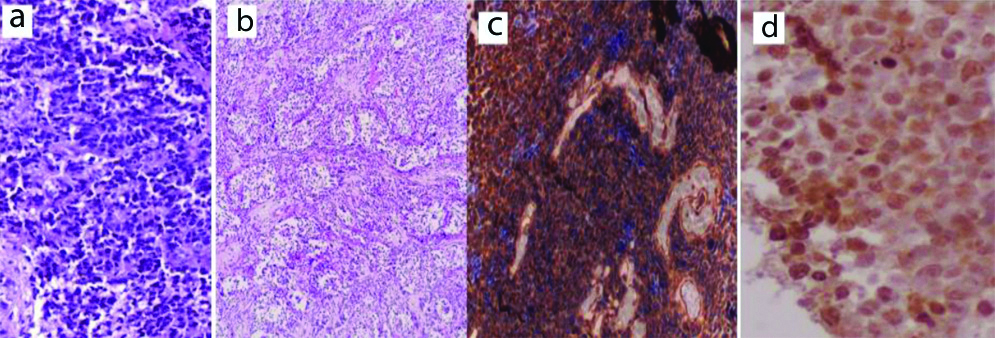
Seven posterior fossa tumours showed solid sheets and diffuse infiltration by undifferentiated round blue cells with frequent mitosis and necrosis. Five cases were classical Medulloblastomas (including one anaplastic variant) and two Desmoplastic Medulloblastomas. Accept one, were negative for β-catenin, and four were positive for Synaptophysin [Table/Fig-18].
Medulloblastoma morphology: a) Classical medulloblastoma (H&E; X200); b) Desmoplastic medulloblastoma (H&E; X100); c) Positive for synaptophysin (IHC; X100); d) Positive for β-catenin (IHC, X400).
Retinoblastoma
Four intra-ocular cases showed solid sheets of small blue cells with hyperchromatic nuclei and scant cytoplasm forming pseudorosettes with frequent mitosis and extensive necrosis. One of them involved the optic nerve. The tumour cells were positive for neuron specific enolase, synaptophysin [Table/Fig-19] and Leu7.
Retinoblastoma morphology; a) H&E (X100); b) Positive for Synaptophysin (IHC, X100).
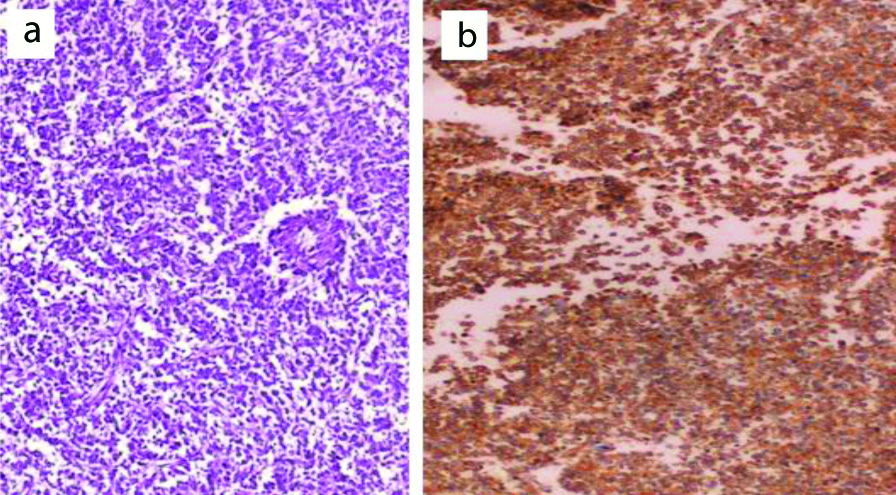
Glioblastoma
Three cases showed infiltrative high grade malignant tumour composed of undifferentiated round cells with evidence of necrosis and complex vascular endothelial proliferation. The background was glial with the tumour cells, expressed a strong GFAP immunostaining [Table/Fig-20].
GFAP positive glioblastoma around complex vascular proliferation (IHC: a, X200, b, X400).
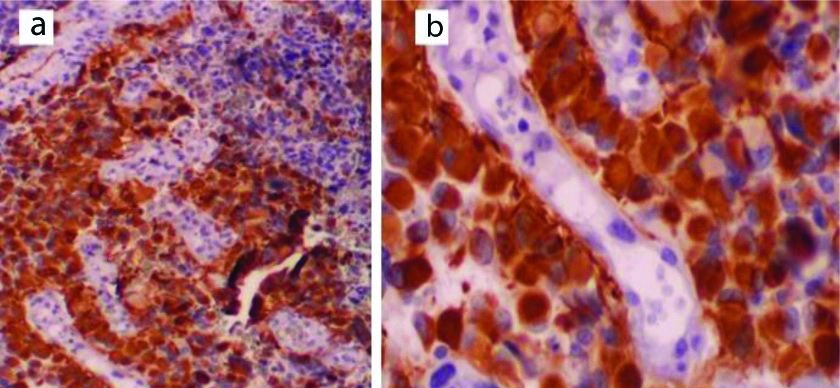
Ependymoma
The term “Ependymoma” was given for 3/120 cases presented as small, bland looking cells within fibrillary background. Pseudorosettes were prominent. Immunohistochemically, the cells expressed GFAP [Table/Fig-21], Statining was positive for Vimentin and focally for EMA and CD99.
Ependymoma morphology: a) H&E (X100); b) IHC positive for Vimentin (X400).
Discussion
Malignant Blue Cell Tumours (MBCT) and Clinical Data
In this study, most patients of MBCT (males and females) were older children (between 11-15 years) with a mean age of 8.49 years. No significant age and sex distribution was found. A little bit younger mean ages (7.4 years) were reported in Islamabad and India [1,13]. Soft tissues formed the commonest location (30%) followed by brain (14.2%), bone and kidney (10% each), then LN (9.2%) and gastrointestinal tract (6.7%). Uncommon sites included adrenals and bone marrow. A previous study in the same region on undifferentiated malignant tumours reported respiratory tract (23.6%) as the most common location followed by gastrointestinal tract [4]. This mismatch can be explained by the fact that that the present cases were confined to the children whereas those of the previous study included all ages with predominant adult and elderly where a respiratory tract usually forms a common site of primary and metastatic undifferentiated carcinoma that appears round cells while adult sarcomas that usually settle in the soft tissues appear spindle rather than round blue.
Malignant Blue Cell Tumours (MBCT) Characterisation
After application of immunohistochemistry, Lymphoma/leukaemia ranked the first among all the identified MBCT (30%), of these 25% were NHL just similar to what has been reported by Pity IS in the same region [4] and Bharathi YK among Indian children [14]. Ewing sarcoma/PNET formed the second most common pathology (24.2%), followed by Neuroblastoma (13%), Wilm’s tumour (9%), Rhabdomyosarcoma (7%) and Medulloblastoma (6%). Few cases were reported as Retinoblastoma, Glioblastoma, Ependymoma, Hepatoblastoma and Astroblastoma. Variable results have been reported among different geographic studies. Bharathi YK, worked on 56 cases in Guntur, India among children up to 14 years; reported that Wilm’s tumour (27%) as the second frequent category, then Rhabdomyosarcoma 14% and Neuroblastoma [14]. Asim M et al., reported NHL (18%) as the second commonest after Wilm’s tumour (20%), then Rhabdomyosarcoma (6%) and Neuroblastoma [13]. Joshi MR et al., in their study among 100 Indians including 76 cases below 14 years, reported ES/PNET (36%) as the most common MBCT in paediatric age groups followed by Neuroblastoma (21%) then NHL [15]. Bansal C et al., reported Retinoblastoma (40.9%) as the dominant neoplasm followed by Neuroblastoma (22.7%) while NHL was the least common entity [1]. Actually, sample size and geographic variations may explain such diverse results.
Malignant Blue Cell Tumours (MBCT) and Final Diagnoses
Lymphomas/Leukaemias
Despite the fact that males formed 70% of Lymphomas/leukaemias and majority of cases presented at 11-15 year ages, no significant age and sex distribution was observed. Most Lymphomas/leukaemias (72.2%) were described at extranodal tissues whereas only 27.8% were nodal at time of diagnosis. Soft tissues was at the top of extranodal positions (25%) followed by GIT (22.2%) and bone marrow (11.1%). B-cell NHL formed the commonest Lymphomas/leukaemias (72.2%), while T-cell lineage was described in 11.1%. The remaining 16.7% cases were Leukaemias. Similarly, Pity IS who studied 127 cases of undifferentiated malignant cell tumours among different age groups, 71.4% of NHL were extranodal and 28.6% were nodal. In the same line, B-cell lineage dominated NHL cases with a minority of T-cell lymphoma [4]. In parallel, Shah SH et al., who studied 61 cases of NHL among Pakistan children reported male dominance and more than half cases were extranodal [16]. In present study, 14 (46.7%) were stratified as DLBL, 26.7% Burkitt’s lymphoma and 13.3% B-cell lymphoblastic lymphoma. In Shah SH et al., study, Burkitt’s lymphoma (33%) was at the top of their NHL followed by Lymphoblastic (28%), then diffuse large cell (15%) [16].
Definite subcategorisation of B-cell NHL is crucial for treatment decision and prognostic stratification. Immunohistochemically, almost all B-cell lineage showed CD45 positivity with at least two of B-cell markers (CD20, PAX5 and CD79a). More than three-fourth of cases were Bcl2 positive with a high mitotic activity (>90% proliferative index). CD10 was positive in 57.1% while Bcl6 and MUM1 were positive in 42.9% each. Given CD10 positivity in consideration with or without Bcl6, 57.1% of DLBL was of germinal centre origin [17]. Proper subcategorisation of CD10+ B-cell lymphoma has become increasingly complex but crucial for therapeutic and prognostic purposes as CD10 positivity harbors a negative impact on treatment response and overall survival [18]. This cell surface glycoprotein can be expressed in a wide variety of cell lineages, including normal germinal centre cells and neoplastic conditions as in the vast majority of follicular lymphoma and Burkitt’s lymphomas. High percentages of acute lymphoblastic leukaemias and a subset of DLBLs also express CD10 [18,19].
In the ongoing study, diagnosis of Burkitt’s lymphoma was given for eight B-cell lineage cases relying on CD10/Bcl6 positivity and 100% proliferative index (Ki67) of tumour cells with absence of Bcl2 adding to their morphologic criteria. It is worth mentioning that other four cases of small B-cell lineage with CD10 positivity and starry sky background were stratified as B-cell lymphoblastic lymphoma. The cells also expressed at least one of TDT and CD99. A single, TDT+/CD99+, case was CD45 negative. As well Fli-1 was positive in a single, CD45-positive, T-LBL. Four T-cell lineage cases with TDT+ and/or CD99+, but CD10 negative, were addressed as T-cell lymphoblastic lymphoma despite negative CD45 in two cases. CD45 negative lymphoblastic lymphomas have been reported [20]. It is interesting to mention that CD99 and Fli-1 positivity with CD45 negativity raised the possibility of Ewing’s/PNET which also represent solid MBCT and are well known to harbor Fli-1 gene mutation. However, TDT positivity adding to Vimentin and PAS negativity strengthened the diagnosis of Lymphoblastic lymphoma. Clinicoradiological, histological and immunophenotypic acumens form the basis for proper diagnosis of LBL and blue cell sarcomas with aberrant immunoprofile.
Ewing Sarcoma/PNET
Despite the fact that definite diagnosis of Ewing’s/PNET necessitates identification of specific chromosomal translocations [21], in this study 29 (about 24%) MBCT with CD99 were labelled as Ewing’s/PNET. Actually, CD99 expression gives a wide carcinomatous, lymphomatous and sarcomatous differential diagnosis [20]. However, diagnosis was corroborated with the available clinical and morphological features in addition to the immunoprofile of tumour cells (Vimentin+, CD99+, Fli-1+) in addition to PAS positivity/PASD negativity with lacking CD45 and TDT immunoreactivity. An interesting finding in the ongoing study was EMA positivity in 41.4% of cases. Indeed epithelial marker expression, like EMA, doesn’t rule out the diagnosis of Ewing’s/PNET [22].
Neuroblastoma
Cytogenetic profile is mandatory for diagnosis of Neuroblastoma. However, given a combined positivity for at least of two endocrine markers (Synaptophysin, NSE, CD56 and Chromogranin) with the traditional morphology, 16 (about 13%) cases were put under the diagnosis of Neuroblastomas despite unusual locations in six cases. Rising urine levels of Vanillylmandelic acid supported the diagnosis [23].
Wilm’s Tumour
Final diagnosis of Wilm’s tumour is through identification of chromosome 11 alterations. However, 11 (about 9%) cases of Wilm’s tumour were reported which presented as undifferentiated small blastemal element. Their nuclei strongly stained for WT1 which is considered a specific marker for Wilm’s tumour [24]. Demonstration of epithelial foci in some cases with negativity for CD45, endocrine and epithelial markers adding to PAS negativity assisted the diagnosis. A worth noting that one case was diagnosed as Wilm’s tumour in a remote site (lung). A striking point in this cases was aberrant strong CD99 immunoexpression. Actually speaking, a previous nephrectomy for Wilm’s tumours, WT-1 cell positivity in addition to their negativity for epithelial, lymphomatous and sarcomatous markers addressed the cases as metastatic Wilm’s tumour [24].
Rhabdomyosarcoma
Alveolar Rhabdomyosarcoma often harbors the typical translocation but Embryonal Rhabdomyosarcoma lacks any specific genetic aberration. Nine (7.5%) MBCT cases were typed as Rhabdomyosarcoma, seven Embryonal and two Alveolar. The cells expressed Vimentin, Desmin, Myogenin, MyoD1 and Myoglobin. Only two cases were stained for CD99. However, clinical, radiological and morphological criteria in addition to the mentioned immunophenotype with negativity for Pankeratin stratified them within the Rhabdomyosarcomas rather than rhabdoid tumour which also expresses Desmin, MyoD1 and Myogenin [25].
Medulloblastoma
Seven (about 6%) posterior fossa brain tumours were assigned as Medulloblastomas including a metastasizing case to the bone. One case showed anaplastic features under the light microscope. In fact, precise diagnosis of Medulloblastoma requires integration of clinicoradiological and histological features with targeted molecular/genetic information. β-catenin positivity allows a precise assignment of patient’s treatment refinement [26].
Retinoblastoma
Four (about 3%) intra-ocular cases were assigned as retinoblastoma relying on the location and traditional histology as well as their neural immunoprofile (i.e., positivity for Neuron specific enolase, Synaptophysin and Leu7 and negativity for CD45, Desmin and GFAP). Such neural immunoprofile was reported by Orellana ME et al., on 39 Retinoblastoma cases in Brazil [27].
Glioblastomas and Ependymoma
There were six GFAP positive brain blue cell tumours within glial background, three showed complex endothelial proliferation and necrosis and reported as Glioblastomas. The other three cases exhibited pseudorosettes around glial neuropils were addressed as Ependymomas. These ependymal cases also expressed EMA and CD99. The last two markers are known to differentiate ependymal from nonependymal brain tumours [28].
Limitation(s)
This was a single town, small-sized, study and these results may not directly reflects other areas in Iraq. Recently discovered highly specific immunomarkers and molecular tests were unavailable so couldn’t be used due to financial purposes.
Conclusion(s)
Integration of clinical data and, histomorphologic acumen eases the categorisation of MBCT. Application of immunohistochemistry in this study has shown a significant improvement in the diagnostic accuracy of paediatric MBCTs. Loss of some differentiation antigens and aberrant expression of some markers often necessitate the use of panels of antibodies and, even molecular testing to target the task.
DLBL: Diffuse large B-cell lymphoma; CD: Cluster of differentiation, FLI-1: Friend leukemia integration 1 transcription factor, NSE: Neuron specific enolase, WT1: Wilm’s tumour 1, PAX: Paired box, TDT: Terminal deoxynucleotidyl transferase, BCL: B-cell lymphoma, Ki67: Proliferative index, GFAP: Glial fibrillary acidic protein and EMA: Epithelial membrane antigen