The ecological community of microorganisms living within the human host is referred to as microbiota and is a mixture of symbiotic, commensal and pathogenic microorganisms. Oral microbiome is defined as the collective genome of microorganisms that reside in the oral cavity. Dysbiosis of the oral microbiota has been linked to several diseases in human beings [1-3]. Some of the oral bacteria are known to be capable of triggering inflammatory processes leading to initiation and progression of Cardiovascular Disease (CVD) indicating the existence of a dynamic relationship between inflammation and the oral microbiome. Oral microbiota has been reported to contribute to the development of atherosclerosis [4].
Oral health promotion has resulted in improvements in periodontal health, and modifications of systemic inflammatory markers [5]. Many of the members of the microbiota are fastidious or cannot be cultured and the advent of culture-independent methods has greatly improved their detection. The diversity and composition of the microbial communities in the oral cavity is studied by metagenomic profiling of the collective genomes of whole microbial communities and modern molecular techniques such as high-throughput sequencing [6]. The 16S rRNA gene which is present in all prokaryotes is approximately 1600 base pairs long and includes nine hypervariable regions (V1-V9). Partial sequences of the gene targeting the hypervariable regions are used for bacterial identification. In recent years, analysis of 16S ribosomal RNA sequences have been used for the taxonomic identification of bacterial strains [7]. There are very few studies on CAD patients, particularly from India. This study reported the comparison of the oral microbiome in patients with CAD and healthy individuals by metagenomic analysis of V3-V4 region of the 16SrRNA gene sequences.
Materials and Methods
A cross-sectional pilot study was carried out between November 2018 and July 2019, in a tertiary hospital specialising in cardiac care in Chennai city, Tamil Nadu, India. The study was approved by the Institutional Ethics Committee (EC Reg no: ECR/140/Inst/TN/2013/RR-16). Subjects of the study were nine adults with coronary angiographic characteristics consistent with significant CAD. Informed consent was obtained from all the patients. Patients admitted for acute myocardial infarction, arrhythmias, cardiac failure or treatment of cardiomyopathies were excluded from the study. One healthy subject with no history of cardiac disease was included as control.
Oral mouthwash samples in phosphate buffered saline were collected in sterile containers. Culture was done on blood agar, MacConkey agar and Mitis salivarius agar. Plates were incubated at 37°C for 24-48 hours after which colonies were identified by standard procedures [8]. Samples were stored at +4° C for metagenomics analysis. DNA was extracted and quantitated. PCR was done using region specific primers to amplify V3–V4 region of 16S rRNA gene [9]. The primers used were:
Forward primer: 5’- CCTACGGGGNGGCWGCAG- 3’
Reverse primer: 5’-GACTACHVGGGTATCTAATCC-3’
Illumina adapter overhang nucleotide sequences were added to the gene specific sequences. The amplicon size was 465 bp. PCR amplicons were analysed on 1.2% agarose gel. Further the libraries were normalised and pooled for sequencing. Sequencing of the variable V3 and V4 regions of the 16S rRNA gene was done using Illumina platform (Genotypic Technology Pvt., Ltd., Bangalore, India).
Statistical Analysis
The paired end V3-V4 reads were checked using FastQC [10] and joined using Fastq-join. Analysis was done using Quantitative Insight into Microbial Ecology (QIIME) [11]. QIIME uses the Ribosomal Database Project (RDP) classifier to assign taxonomic data to each representative sequence using a cut-off of ≥97% sequence similarity against the reference database. Relative abundance was calculated using the R package Non-negative Matrix Factorization (NMF).
Results
The nine patients (7 males, 2 females) aged 45-76 years and were included from the tertiary hospital patients. The clinical and demographic details of the patients are given in [Table/Fig-1]. All specimens grew abundant normal flora, predominantly Viridans Group Streptococci (VGS). Among the VGS, Streptococcus salivarius and Streptococcus mitis were most frequently isolated in patients while the control sample grew S. mitis. When the V3-V4 region of 16S rRNA gene were analysed, it was seen that Firmicutes and Bacteroidetes were the predominant phyla and accounted for more than 60% of the taxa in all the patients. Proteobacteria was the predominant phylum in the control and together with Firmicutes accounted for more than 60% of the taxa. Eighteen bacterial phyla were identified within the combined dataset [Table/Fig-2]. In 8/9 patient samples and in the control sample, the six major phyla, Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and Fusobacteria, contained 99% of the taxa and the remaining phyla, Tenericutes, TM7, Synergistetes, Planctomycetes, Verrucomicrobia, SR1, Chloroflexi, etc., contained less than 1% of the taxa. Phyla such as Verrucomicrobia which was present in the control was very rare in CAD patients (1/9). Cyanobacteria and Chlorobi were absent in the control and rare in CAD patients.
Demographic details and clinical history of patients.
| S. No. | Sample ID | Age | Sex | Height (cm) | Weight (kg) | Clinical history | Diabetic status | Lipid profile | Culture results |
|---|
| 1 | CAD 11 | 61 | Female | 158 | 56 | TVD-s/p OPCAB CAG done on 21/11/18 | Diabetic | Not available | Abundant S.mitis, occasional S. salivarius |
| 2 | CAD 12 | 64 | Male | 162 | 61.3 | TVD-CAG done on 16/11/18 | Diabetic | TC-96, TGL-121, HDL-32, LDL -62 | Gram negative diplococci, S. mitis, S. salivarius, S.mutans |
| 3 | CAD 13 | 63 | Male | 150 | 65 | SVD CAG done on 5/12/18 | Diabetic | TC-100, TGL-188, HDL-30, LDL-66 | Gram negative diplococci, abundant S.mitis, few S. salivarius, very few S. mutans, occasional Entercocci |
| 4 | CAD 14 | 65 | Male | 160 | 87.5 | TVD-CAG done on 1/12/18 | Diabetic | TC-197, TGL-189, HDL-33, LDL-136 | Abundant S.mitis, S.epidermidis |
| 5 | CAD 15 | 53 | Female | 161 | 68 | SVD, PTCA-stent on 5/12/18 | Diabetic | TC-116, TGL-107, HDL-35, LDL-75 | Abundant S.mitis, scanty Entercocci |
| 6 | CAD 17 | 76 | Male | 160 | 51.7 | TVD, CAG done on 30/11/18 | Diabetic | TC -173, TGL -163, HDL -24, LDL-143 | Micrococci, gram negative diplococci, S. mitis, occasional Entercocci |
| 7 | CAD 18 | 51 | Male | 172 | 63 | TVD, CAG done on 12/11/18 | Diabetic | TC-111, TGL-173, HDL-34, LDL-67 | Abundant S. mitis, S. salivarius |
| 8 | CAD 20 | 46 | Male | 166 | 78.5 | TVD- CAG done on 14/11/18 | Diabetic | TC-137, TGL- 223, HDL-30, LDL-89 | S. mitis, S. salivarius, few Enterococci. |
| 9 | CAD 21 | 49 | Male | 160 | 60 | TVD-s/p OPCAB | Diabetic | TC- 162, TGL-166, HDL-27, LDL-117 | S. salivarius |
CAD: Coronary artery disease; TVD: Triple vessel disease; SVD: Single vessel disease; CAG: Coronary angiography; s/p: Status post-surgery; PTCA: Percutaneous transluminal coronary angioplasty; OPCAB: Off pump coronary artery bypass; TC: Total cholesterol; LDL: Low-density lipoprotein; HDL: High-density lipoprotein; TGL: Triglycerides
| Patient ID | CAD11 | CAD12 | CAD13 | CAD14 | CAD15 | CAD17 | CAD18 | CAD20 | CAD21 | Control |
|---|
| Clinical findings | TVD | TVD | SVD | TVD | SVD | TVD | TVD | TVD | TVD | |
| Age/Sex | 61/F | 64/M | 63/M | 65/M | 53/F | 76/M | 51/M | 46/M | 49/M | 32/F |
| Phylum |
| Firmicutes | 45.9381 | 53.2765 | 34.6035 | 75.4709 | 37.643 | 69.996 | 64.8542 | 66.9725 | 70.9569 | 30.9947 |
| Bacteroidetes | 26.5174 | 17.7554 | 28.6291 | 7.7481 | 24.5752 | 13.5547 | 16.5183 | 10.1216 | 16.8357 | 19.158 |
| Actinobacteria | 8.9271 | 6.2999 | 2.296 | 1.0211 | 5.4395 | 0.9515 | 10.1754 | 6.7335 | 7.5231 | 3.7984 |
| Proteobacteria | 8.2379 | 11.2332 | 13.8162 | 12.172 | 22.0904 | 10.3896 | 5.4761 | 15.9481 | 2.2843 | 34.9574 |
| Fusobacteria | 7.3987 | 10.3626 | 9.9435 | 3.1587 | 9.6968 | 3.9489 | 2.2326 | 0.1787 | 1.9498 | 9.3998 |
| Spirochaetes | 2.1427 | 0.0864 | 6.6048 | 0.2048 | 0.0442 | 0.7344 | 0.1762 | 0.0187 | 0.0421 | 0.996 |
| Tenericutes | 0.4046 | 0.0638 | 1.4877 | 0.0098 | 0.0031 | 0.1742 | 0.009 | 0.0021 | 0.0444 | 0.0371 |
| TM7 | 0.2397 | 0.7389 | 1.1436 | 0.1711 | 0.3859 | 0.0884 | 0.5003 | 0.0083 | 0.2187 | 0.5245 |
| Synergistetes | 0.1568 | 0.07 | 1.3571 | 0.0084 | 0.0244 | 0.1274 | 0.0566 | 0.0021 | 0.1158 | 0.0106 |
| Planctomycetes | 0.0288 | 0.0412 | 0.0044 | 0.0084 | 0.0031 | 0.013 | 0 | 0.0021 | 0.0152 | 0.0353 |
| Verrucomicrobia | 0.0046 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0018 |
| Thermi | 0.0023 | 0 | 0 | 0 | 0 | 0 | 0 | 0.0125 | 0.014 | 0 |
| Cyanobacteria | 0.0012 | 0.0041 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SR1 | 0 | 0.0123 | 0.0756 | 0.0266 | 0.093 | 0 | 0.0013 | 0 | 0 | 0.0406 |
| GN02 | 0 | 0 | 0 | 0 | 0.0015 | 0 | 0 | 0 | 0 | 0 |
| Chloroflexi | 0 | 0.0268 | 0.0267 | 0 | 0 | 0.0182 | 0 | 0 | 0 | 0 |
| Chlorobi | 0 | 0 | 0.0104 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Acidobacteria | 0 | 0.0288 | 0.0015 | 0 | 0 | 0.0039 | 0 | 0 | 0 | 0.0459 |
TVD: Triple vessel disease; SVD: Single vessel disease
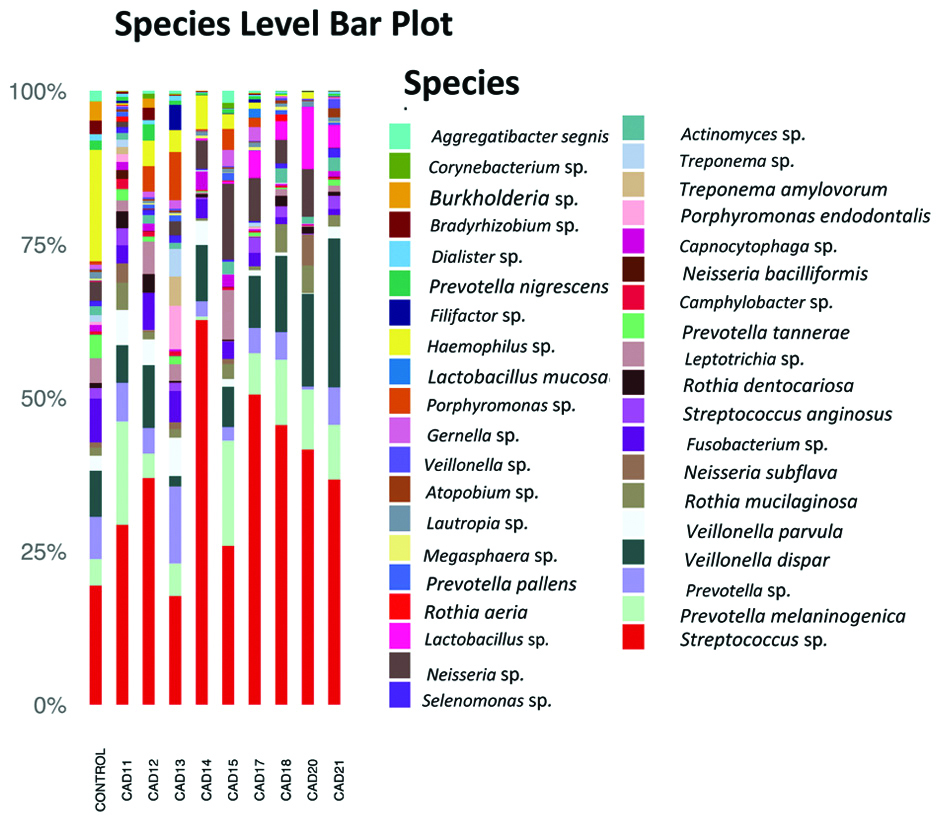
In the single control sample, Proteobacteria (34.9%), Firmicutes (31.99%), and Bacteroidetes (19.1%) were the most abundant. In the patients, Firmicutes varied between 35% and 75%, Proteobacteria, from 2.2% to 22.0%, and Bacteroidetes from 7.7% to 28.6%. The most common genus was Streptococcus in all the samples, followed by Prevotella and Veillonella [Table/Fig-3].
Relative abundance of species of the oral communities from 9 patients and one healthy individual.
Discussion
Previous studies on the composition of the oral microbiota in healthy individuals and in patients with atherosclerosis have shown that the oral microbiota of patients is dominated by Firmicutes (69%) followed by Bacteroidetes (10%), Actinobacteria (9%), Fusobacteria (6%), Proteobacteria (5%), and <1% of Spirochaetes, TM7, SR1, and Tenericutes. The microbiome in healthy individuals was found to be similar except for more representation from Firmicutes (76%) and less of Bacteroidetes (6%) and Fusobacteria (3%) [12]. Other studies of the oral microbiome in healthy individuals have shown that Firmicutes though most abundant accounted for only 33.2% of all sequences, Proteobacteria for 27.5%, Bacteroidetes for 16.6% and Actinobacteria for 14.5% of all sequences [13].
In the present study, Firmicutes, Proteobacteria and Bacteroidetes were the most abundant phyla in all the patient samples with large inter-sample variations. The phylum Proteobacteria was more abundant in the control sample than in any of the patient samples. Though only one control sample was analysed in this study, the abundance of Firmicutes, Proteobacteria and Bacteroidetes was comparable to a study done by Bik EM et al., on 10 healthy individuals [13]. It was also comparable to author’s earlier study which investigated the oral microbiome in four healthy individuals, according to which Bacteroidetes, Proteobacteria and Firmicutes were the predominant phyla seen in oral cavity of healthy individuals. There was considerable diversity in the microbiota and uncultured bacteria showed the highest abundance [14]. In the present study Fusobacteria ranged between 0.1% to 10.3% in patients and 9.3% in the control. It appeared that the abundance of Fusobacteria varied widely in the patient population. Other studies have reported 6% in patients [12] and 3%-6.7% in controls [12,13]. The abundance of Fusobacterium is said to positively correlate with levels of LDL cholesterol [12], but no such association was seen in the present study. Streptococcus spp., and Veillonella spp., were abundant in the patient samples which was comparable to a previous study [15]. Both organisms have previously been isolated from atherosclerotic plaques and plaque microbiota are said to be derived from the oral cavity and/or the gut.
Limitation(s)
This was a pilot study and the sample size of this study was small. The high cost of sequencing is restrictive and pilot studies help to determine statistical power and optimum sample size [16]. Metagenomics research generates large number of genome sequences and consumes heavy computing resources and time. Sample sizes are kept to an optimum, particularly in low resource settings to avoid redundancy and reduce costs.
Conclusion(s)
Advances in molecular biology and bioinformatics have made microbiome research possible. A 16SrRNA gene sequencing is superior to culture methods for studying abundance and diversity of the oral microbiome. Microbial communities in the oral cavity varied considerably between individuals and there was no overall difference when the oral microbiome in CAD patients was compared to the normal oral microbiome.
CAD: Coronary artery disease; TVD: Triple vessel disease; SVD: Single vessel disease; CAG: Coronary angiography; s/p: Status post-surgery; PTCA: Percutaneous transluminal coronary angioplasty; OPCAB: Off pump coronary artery bypass; TC: Total cholesterol; LDL: Low-density lipoprotein; HDL: High-density lipoprotein; TGL: Triglycerides
TVD: Triple vessel disease; SVD: Single vessel disease