Neonatal Autopsies with Heart Disease: A Challenge for Pathologist
Smita Singh1, Srijan Srivastav2, Kiran Agarwal3
1 Professor, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
2 Senior Resident, Department of Pathology, Lady Hardinge Medical College, Noida, Uttar Pradesh, India.
3 Director Professor, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Srijan Srivastav, A 537, Sector 47, Noida, Uttar Pradesh, India.
E-mail: dr.srijansrivastav@gmail.com
Congenital Heart Diseases (CHD) carry significant morbidity and mortality in paediatric patients. Transposition of Great Arteries (TGA) is a common cyanotic CHD. However, onset of Pulmonary Arterial Hypertension (PAH) is potentially severe and rare complication reported in 1-3% of newborns. Hypertrophic Cardiomyopathy (HOCM) is a primary disease of cardiac muscle usually recognised in adulthood. Neonatal HOCM without other congenital anomalies, no family history of HOCM, no history of exposure to corticosteroids or any inborn errors of metabolism is rarely recognised. Authors hereby report two cases of neonatal autopsy showing TGA with PAH (Grade 2) and another with HOCM without any primary cause received in our hospital. With this case report authors wish to stress on the importance of cardiac examination (heart and the associated vessels) in autopsy specimen which in turn requires training of pathologists in cardiac pathology along with routine fetal anomaly scanning in arriving successively at the final diagnosis and detecting the cause of death which helps in management of future pregnancies by the clinicians.
Congenital anomaly, Hypertrophic cardiomyopathy, Transposition of great arteries
Case Reports
Case 1
A 3 kg female baby was delivered and cried immediately after birth but thereafter showed poor respiratory efforts. On examination, baby was cold and cyanosed, heart rate was 108/minute, SPO2 was 34% and capillary refill time was >3 seconds. Peripheral pulses were not palpable while central pulses were feeble. Baby was intubated and kept on ventilator support. After one hour of failed respiratory efforts, the baby expired.
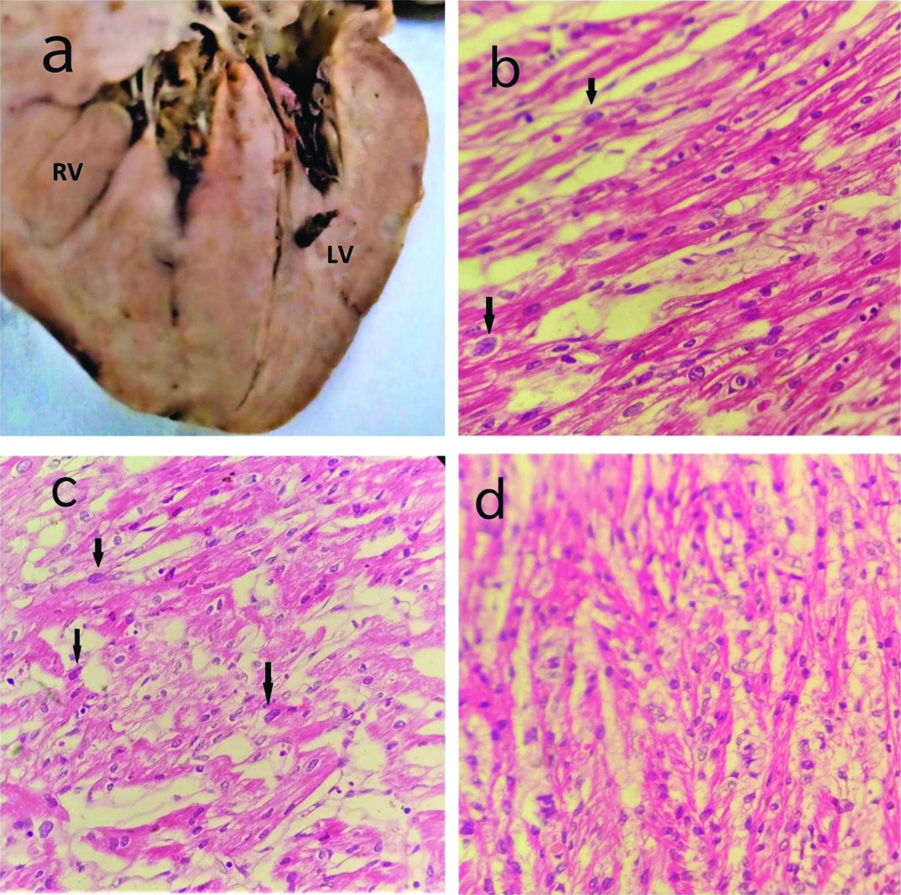
At autopsy, all the organs were found to be congested. On gross examination, heart measured 4.5×3.5×2.5 cm and weighed 19 grams [Table/Fig-1a]. On opening by in-flow outflow method, aorta was seen arising from right ventricle and measured 0.6 cm in diameter, pulmonary trunk was seen arising from left ventricle and measured 0.5 cm in diameter. Gross hepatomegaly was seen. Liver measured 10.5×6.0×4.5 cm and weighed 166 grams. No other significant findings were seen in any other organ. Microscopic examination of heart showed mild congestion in all the chambers of heart. Sections from pulmonary artery showed medial hypertrophy with mild fibrous hypertrophy in the intimal layer [Table/Fig-1b-d]. Microscopic features were suggestive of TGA with pulmonary artery hypertension. Microscopic sections from liver showed mild congestion with extramedullary hematopoiesis.
(a) Gross photograph of heart showing pulmonary artery arising from left ventricle; (b) Micro photograph showing aorta arising from right ventricle; (c) Photomicrograph showing section from pulmonary artery showing medial hypertrophy (H&E,100X); (d) Photomicrograph showing section from pulmonary artery showing medial hypertrophy with mild fibrosis in the intima (H&E,400X).
RV: Right ventricle; LV: Left ventricle
Case 2
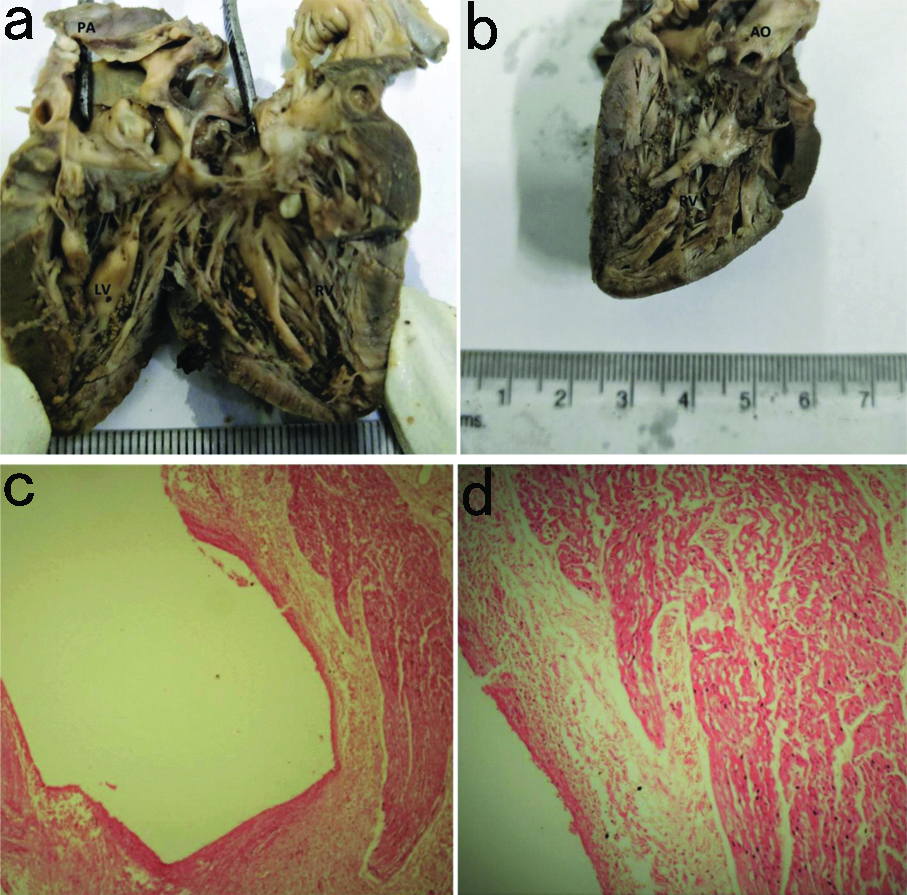
A neonate delivered through normal vaginal delivery died one week after birth allegedly of unknown cause. At autopsy, gross examination of heart, lungs, kidney, spleen and liver showed congestion. Heart measured 5×3×2.2 cm and weighed 22 grams, both the ventricles appeared hypertrophied with free wall of left and right ventricle measuring 1.1 cm and 0.5 cm, respectively [Table/Fig-2a,b]. Interventricular septum was thickened. Microscopic examination of section from right and left atrium showed congestion. Sections from left and right ventricles showed areas of muscle fibre disarray and myofibre hypertrophy with nuclear enlargement with focal area of fibrosis, suggestive of HOCM [Table/Fig-2c,d].
(a-b) Gross photograph of fetal heart showing biventricular hypertrophy (RV>LV); (c-d) Photomicrograph showing muscle disarray, hyperchromatic nuclei (black arrows) and areas of fibrosis (H&E; 400X).
PA: Pulmonary artery; LV: Left ventricle; RV: Right ventricle; AO: Aorta
Discussion
Heart develops from a single tube into a four-chambered structure via a complex series of loopings and septations. Given the complexity of cardiac development, congenital cardiac defects account for most cardiac diseases in childhood. However, TGA is one of the most common cyanotic CHD accounting for 5-7% of all patients with CHD [1]. Onset of PAH in patients with TGA is a potentially severe and rare complication which is represented in our first case. TGA in the newborn combined with PAH was reported previously to occur in 1-2 cases per million inhabitants [1].
The embryological defect in TGA stems from abnormal formation of truncal and aortopulmonary septa. PAH results from failure of pulmonary circulation to dilate at birth. It is characterised by persistent elevation of pulmonary vascular resistance. Abnormal pulmonary vasoreactivity to vasoconstrictor and vasodilator stimuli is involved in pathogenesis [2]. Our first case showed TGA associated with PAH. This case showed grade 2 changes (in Heath and Edwards classification) in pulmonary vasculature [3]. Intimal proliferation in association with Transposition of the Great Arteries and intact ventricular septum is rarely reported in infants [4-6]. Compared with patients with other PAH aetiologies, the increases in pulmonary pressure seen in patients with PAH-CHD occur early.
Left to right shunts like PDA, increase pulmonary blood flow, but are not initially associated with cyanosis unlike TGA. However, these shunts chronically elevate both volume and pressure in the normally low pressure, low resistance pulmonary circulation. Initially muscular pulmonary arteries with size less than 1 mm undergoes medial hypertrophy and vasoconstriction for the maintenance of normal pulmonary capillary and venous pressures. But in the later stages of prolonged pulmonary arterial vasoconstriction there develops irreversible obstructive intimal lesions in the arteries which are comparable to the changes seen in systemic hypertension [7]. But the development of these vascular change in pulmonary vessels is unusual in TGA which being a right to left shunt condition. Some studies suggest that the forces that act upon the lungs in TGA differ from those present in other CHDs.
The onset of PAH in patients with TGA is a potentially severe complication first described decades ago. Mechanism of PAH in TGA is complicated and not well understood. According to Aziz KU et al., the importance of bronchial artery circulation in d-transposition - pulmonary artery anastomosis as a mechanism by which pulmonary artery lesions develop soon after birth [8].
Patent foramen ovale or a patent ductus plays a crucial role in the maintenance of life in infants with TGA and an intact ventricular septum. This small but effective blood flow saturates the blood in pulmonary artery with oxygen and leaving aortic blood unsaturated. Thus, the oxygen needed for maintenance of life is taken up by increased pulmonary blood flow and delivered by increased systemic blood flow. With the increased blood flow through the pulmonary vascular bed, perfusion pressure increases, thereby, leading to increased pulmonary pressures. Findings characteristic of PAH are medial hypertrophy and intimal fibrosis.
Our second case showed HOCM. HOCM in infants may mimic other CHDs, and its clinical features differ often from that observed in HOCM in children and adults [9]. Substantial evidence indicates that HOCM in children and adults is often genetically transmitted [10]. HOCM in children are generally secondary, due to the sequelae of obstructive CHD, or due to inborn errors in metabolism, such as glycogen storage disease or mucopolysaccharidosis or with other CHDs such as ventricular septal defects and tetralogy of fallot [11]. In our case, infant had no family history of HOCM and no history of exposure to corticosteroids or showed any inborn errors in the metabolism.
Microscopically, HOCM is characterised by cardiomyocyte hypertrophy and myofibrillar disarray in greater than 5% of myocardial tissue along with fibrosis. Myofibres are interspersed in different directions and forms whorls around fibrotic area and may even be aligned perpendicular to each other. The disarray also involves intracellular myofibrils and connective tissues which lead to interstitial fibrosis and finally progressing to lumen reduction of microvascular coronary artery walls which results in ischemia and fibrosis, usually seen in areas of greater hypertrophy. The inefficient contractility of sarcomere proteins which activates insulin growth factor and tissue growth factors along with angiotensin II, being the postulated mechanism of disarray and fibrosis [12]. Myocardial disarray may be associated with electrical conduction abnormalities in the heart which contributes to increased incidence of sudden cardiac death in such patients.
Conclusion(s)
Autopsy rates in neonates who died are declining worldwide. Many postmortem ancillary techniques are suggested as an adjunct to or substitute for autopsy. Our cases highlight the potential benefit of introducing cardiac pathology training for pathologists and need of autopsy in all cases to aid in identification of cause of death in neonates.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: May 18, 2020
Manual Googling: Jun 25, 2020
iThenticate Software: Jul 31, 2020 (13%)
[1]. Manzano PD, Soto AM, Barba R, Galdo AM, Izquierdo AG, Pulmonary artert hypertension and neonatal arterial switch surgery for correction of transposition of the great arteriesRevista Espanola De Cardiologia 2016 69(9):836-41.10.1016/j.recesp.2016.01.035 [Google Scholar] [CrossRef]
[2]. Walsh M, Stork E, persistent pulmonary hypertension of the newborn. Rational therapy bases on pathophysiologyClin Perinatol 2001 28(30):609-27.10.1016/S0095-5108(05)70109-3 [Google Scholar] [CrossRef]
[3]. Heath D, Edwards JE, The pathology of hypertensive pulmonary vascular diseaseCirculation 1958 18(4 part 1):533-47.10.1161/01.CIR.18.4.53313573570 [Google Scholar] [CrossRef] [PubMed]
[4]. Villafane J, Lantin-Hermoso MR, Bhatt AB, D-transposition of the great arteries: The current era of the arterial switch operationJ Am Coll Cardiol 2014 64(5):498-511.10.1016/j.jacc.2014.06.115025082585 [Google Scholar] [CrossRef] [PubMed]
[5]. Sarris GE, Balmer C, Bonou P, Comas JV, Cruz ED, Clinical guidelines for the management of patients with transposition of the great arteries with intact ventricular septumEuropean J Cardiothorac Surg 2017 51(1):e1-32.10.1093/ejcts/ezw36028077506 [Google Scholar] [CrossRef] [PubMed]
[6]. Kumar A, Taylor GP, Sandor GGS, Patterson MWH, Pulmonary vascular disease in neonates with transposition of the great arteries and intact ventricular septumBr Heart J 1993 69:442-45.10.1136/hrt.69.5.4428518068 [Google Scholar] [CrossRef] [PubMed]
[7]. Kumar R, Abbas A, Malone E, Robbins and Cotran Pathological basis of disease, the heartElsevier 2015 129th edition:553 [Google Scholar]
[8]. Aziz KU, Paul MH, Rowe RD, Bronchopulmonary. Circulation in d-transposition of the great arteries: Possible role in genesis of accelerated pulmonary vascular diseaseAnn J Cardiol 1977 39:432-81.10.1016/S0002-9149(77)80101-X [Google Scholar] [CrossRef]
[9]. Sankaranarayan RJ, Fleming EJ, Garratt C, Mimics of hypertrophic Cardiomyopathy- diagnostic clues to aid early identification of phenocopiesArrhythm Electrophysiol Rev 2013 2(1):36-40.10.15420/aer.2013.2.1.3626835038 [Google Scholar] [CrossRef] [PubMed]
[10]. Naseer WK, Williams JF, Mishkin ME, Childress RH, Helmen C, Familial myocardial disease with and without obstruction to left ventricular outflow. Clinical, hemodynamics and angiographic findingsCirculation 1967 35:63810.1161/01.CIR.35.4.6386067233 [Google Scholar] [CrossRef] [PubMed]
[11]. Seo HS, Lee IH, Song YW, Choi BM, Jang GY, Son CS, A case of congenital hypertrophic cardiomyopathyKorean Circ J 2013 43(1):54-56.10.4070/kcj.2013.43.1.5423407623 [Google Scholar] [CrossRef] [PubMed]
[12]. Seidman JG, Seidman C, The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigmsCell 2001 104(4):557-67.10.1016/S0092-8674(01)00242-2 [Google Scholar] [CrossRef]