The GCTs of ovary are of sex cord origin and were first described by Rokitansky in 1855. GCT constitutes less than 2% of all the ovarian tumours [1]. Only 0.1% of all ovarian tumours and 4-5% of GCT are reported in children. These tumours may be subdivided into adult type (AGCT-95% of the cases) and juvenile type (JGCT-5% of the cases) based on the histological characteristics and age of onset. JGCT is a subtype of ovarian sex cord stromal tumour with a favourable prognosis if diagnosed at an early stage. The juvenile type of ovarian tumour was first described in 1977 by Scully RE [2]. JGCT is an uncommon tumour and may be associated with endocrinal secretions [3,4]. The clinical presentations associated with the hormonal changes include precocious pseudopuberty, vaginal bleeding, irregular menstruation, growth spurt, rarely virilisation or hirsuitism, abdominal pain and abdominal distention [5]. The prognosis is good in patients diagnosed in Stage I, especially after surgical resection alone. GCT is mostly unilateral in occurrence [6]. Occurrence of JGCT in 11 females less than 15 years was studied extensively.
Juvenile variety of GCT is rare. This study was thus undertaken so as to analyse the clinico-pathological features along with the laboratory findings including the IHC features. The present study helped in analysing the key features of this rare entity.
Materials and Methods
The prospective study was carried out in 11 cases of ovarian JGCT in female children and was evaluated according to the management and outcome. The patients were reported from tertiary care teaching institutions i.e., VCSGG Medical Sciences and Research Institute, Srinagar Garhwal, PauriGarhwal, Doon Medical College, Dehradun and Rohilkhand Medical College, Bareilly. The study duration was three years starting from April 2017 to March 2020. Institutional Ethical Clearence was obtained (IEC/GDMC/105).
Clinico-pathologic features including age, presenting complaints, laboratory findings, radiological features, site of tumour, staging, operative procedure were done, gross and microscopic findings and adjuvant therapy if used were recorded. Fertility sparing surgery was done in 10 cases while, one girl diagnosed in a later stage had to undergo total abdominal radical hysterectomy with bilateral salpingo-oophorectomy. In the laboratory findings besides all hormonal profile of all the 11 patients were studied. All the resected specimens were fixed in 10% buffered formalin. After grossing and processing 3-4 μ sections were cut and routinely stained using H&E staining. IHC markers including Vimentin, Inhibin, CD99, Pancytokeratin (Pan CK) and Low molecular weight CK were applied as per the manufacturer’s guidelines. For staging The International Federation of Gynaecology and Obstetrics (FIGO) 2014 classification was used [1]. All the findings were analysed as per the age of the patient, gross, microscopic findings as well as the expression of different IHC markers.
Results
A total of 11 cases of JGCT were included in the present study. Majority of the (7 cases) females aged between 5-8 years [Table/Fig-1] with no significant past medical history. They presented with the symptoms of enlargement of breast, intermittent vaginal bleeding, and abdominal pain since past few months and development of downy pubic hair. Three cases presented with abdominal distension due to ascites and one case had symptoms of acute abdomen. The tumour was on the right side in eight patients and on the left side in the other three.
Age distribution in Juvenile Granulosa Cell Tumour (JGCT).
| Age group (in years) | Number of cases |
|---|
| 5-8 | 7 |
| 9-12 | 3 |
| ≥13 | 1 |
| Total | 11 |
The serum laboratory findings are shown in [Table/Fig-2]. The total T4 and TSH were in the normal range. The serum Inhibin levels were raised in all the 11 cases. The levels of βHCG, AFP, CA-125, CEA and CA-19.9 were within normal limits.
Levels of hormone as compared with normal range.
| Hormone | Levels in patients | Normal Range |
|---|
| Estradiol | 170-221 | <36 pmol/L |
| Luteinizing Hormone (LH) | <0.3 | 1-18 IU/L |
| Follicular Stimulating Hormone (FSH) | <0.4 | 2-12 IU/L |
| T-3 (Triiodothyronine) | 90-112 | 3-6 pmol/L |
| T-4 (Thyroxine) | In normal range | 5.5-12.5 μg/dL |
| TSH (Thyroid Stimulating Hormone) | In normal range | 0.5-5 mIU/L |
Ultrasonography (USG) of the pelvis showed ovarian multiseptated cystic mass with solid component and normal endometrial and myometrial echo pattern. Chest CT scan and bone scans were unremarkable. Diagnosis was made as JGCT. Out of 11 cases, seven cases were FIGO stage Ia, three cases were stage Ic and one case was stage IIc. In seven cases of stage Ia unilateral salpingo-oopherectomy was done. In three cases of stage Ic unilateral salpingo-oopherectomy followed by radiotherapy and chemotherapy was done. In the single case of stage IIc total abdominal radical hysterectomy with bilateral salpingo-oopherectomy was performed. Follow-up of the patients showed improvement and response to treatment. The five year survival rate in stage Ia and Ic ranged between 80-90%, and in stage IIc it was 70%.
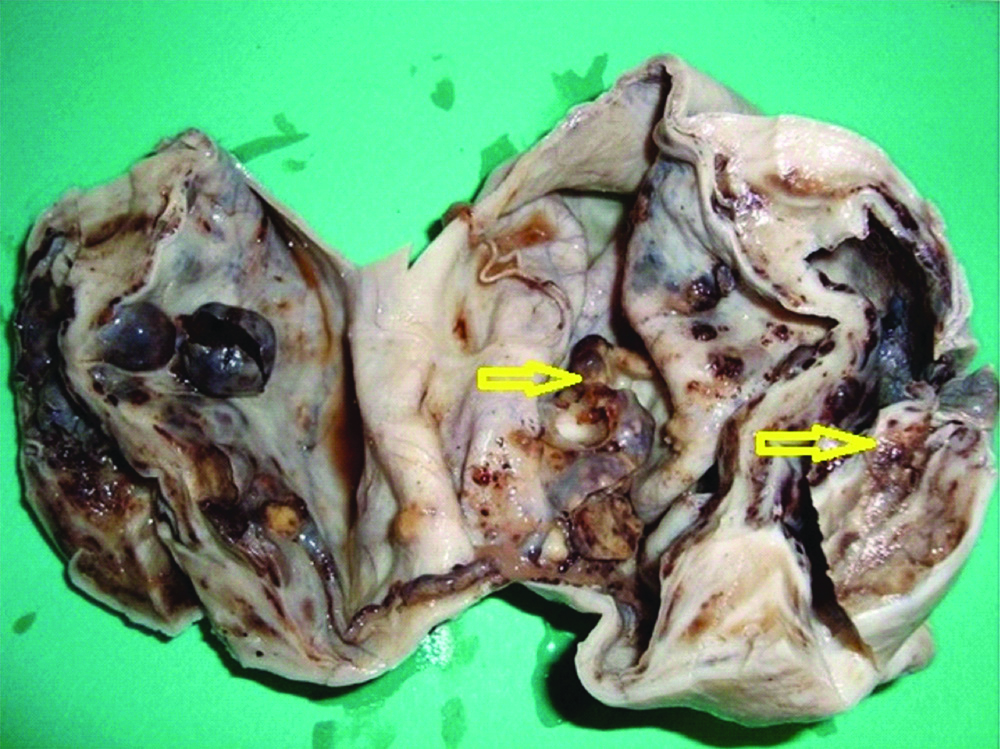
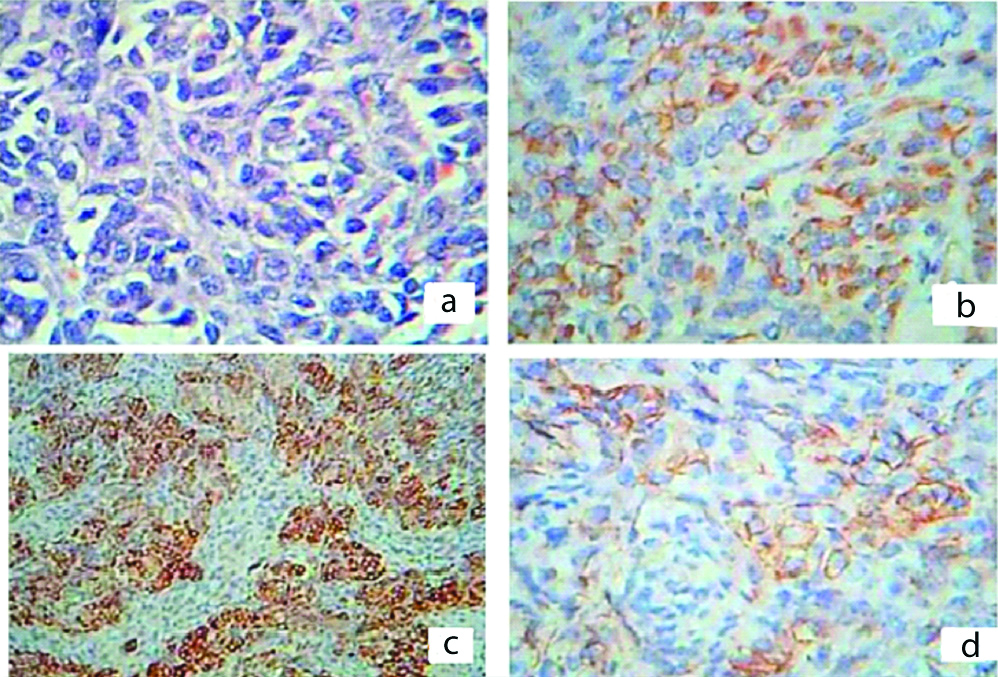
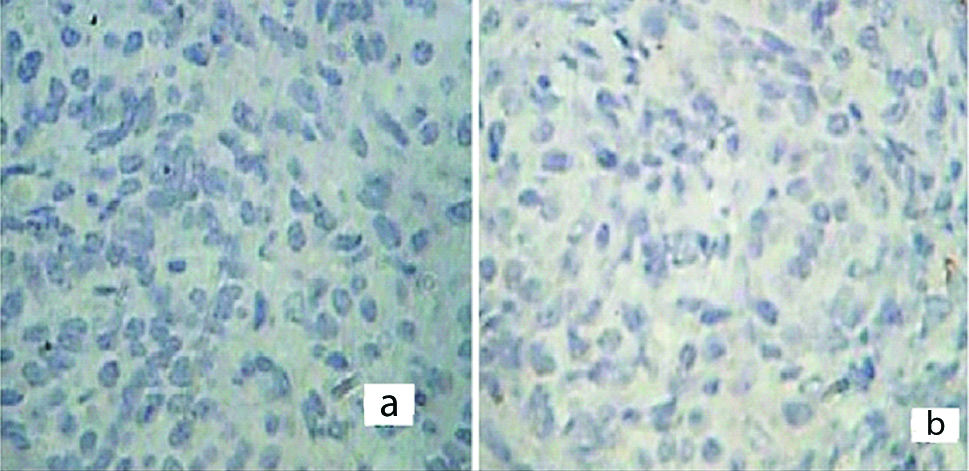
The resected specimens were subjected to histopathological examination. The outer surface of tumour was smooth and showed prominent vascular markings. Cut-section of the tumour showed large predominantly cystic, multiloculated, filled with clear yellow fluid and scattered papillary projections in majority of the tumours [Table/Fig-3]. Histopathology in all the cases revealed ovarian tissue partially replaced by nodules comprising of sheets of ovoid and polygonal pale eosinophilic to clear cells [Table/Fig-4a]. Multiple irregular follicles showing cystic changes were present in three of the 11 cases. Proliferations of granulosa-like cells in the ovarian stroma were also noted in eight patients. Immunohistochemistry of the tumour cells showed immunopositivity for Vimentin, Inhibin and CD99 [Table/Fig-4b-d] and immunonegativity for Pancytokeratin (Pan CK) and Low molecular weight Cytokeratin in all the cases [Table/Fig-5a,b]. The overall final impression of JGCT was made in these patients.
Cut-section of tumour showing multi-loculated cyst with scattered papillary projections (arrow).
a) Histopathology showing sheets of ovoid and polygonal pale eosinophilic to clear cells (H&E x400). b-d) Showing immunohistochemistry of the tumour cells exhibiting positivity for Vimentin (X 400; b), Inhibin (X100, c) and, CD 99 (X400, d).
Immunohistochesmistry of the tumour cells showing immmunonegativity for Pancytokeratin (X400, a) and Low molecular weight Cytokeratin (X400, b).
Discussion
GCT of ovary was first described in 1855 by Rokitansky. Characterised by the clinico-pathological differentiation granulosa cells can be juvenile or adult type. JGCT of ovary was first described by Scully RE in 1977 [2]. JGCT varies from the adult type in the clinical features, histopathological picture as well as in the expression of IHC markers [6]. JGCT comprises of only 0.1% of all ovarian tumours and only 4-5% of all GCTs occur in children [7,8]. JGCT occurs in premenarchal girls and young females, and the mean age at diagnosis is 13 years [9]. Generally, it occurs in premenarchal girls or young women. However, it has been reported in a wide age range from 10-month-old child to a 67-year-old lady [10,11]. They are predominantly composed of granulosa cell, theca cell and fibroblast in different combinations. Sexual precocity in prepubertal girls due to granulosa and theca cell tumour occurs in only 1% of all the cases [12]. The hyperestrogenic state by GCT results in metrorrhagia and amenorrhoea. Increased level of Inhibin leads to FSH suppression resulting in amenorrhoea. Other manifestations include concomitant endometrial hyperplasia and adenocarcinoma and increased chances of breast malignancy later in life. Endometrial adenocarcinoma has also been noted in 5-10% of patients and is often well-differentiated presenting in an early stage with a favourable prognosis [13]. The patients with secretory GCTs mostly present with signs of precocious pseudopuberty like breast development, pubic and axillary hair growth, clitoral hypertrophy, vaginal secretions or discharge, metrorrhagia, physical growth spurt and other secondary sex characteristics [9,14-16]. The most common symptoms are abdominal pain and distension [1,9]. There may be associated Ollier disease, Mafucci syndrome, and abnormal karyotype or ambigious genitilia [9]. In contrast to adult GCT, JGCT has a high mitotic index and more aggressive tumour growth [1].
Primary gonadal disorder, adrenal gland, exogenous secretion of gonadotropin by a tumour or autonomous ovarian cyst must be excluded in these cases [17]. Patients with recurrent ovarian cyst formation and persistence of the cysts especially with a significant solid component beyond three months should alarm the gynaecologist of the possibility of patient having ovarian JGCT [17]. On Computed Tomography (CT) and USG, they appear as large, multiloculated masses, with thin or thick and irregular septations, as well as solid components or large multicystic masses [18]. In all cases in present study, USG of the pelvis showed ovarian cyst with solid component and irregular septations. The most common finding on physical examination noted was a palpable mass [9]. The median diameter of the tumour was 7.5 cm (ranging from 4-16 cm). The tumour was resected completely in all the patients. No tumour was found in the omentum in any of the patients. It has been observed that large tumours greater than 10-15 cm in diameter are known to have a high recurrence rate with a reduced progression-free survival, irrespective of the tumour stage [19]. However, some authors have demonstrated that tumour size did not have any prognostic value as compared to the stage. Recurrence was not noted in any of the patients. Microscopically, there were diffuse and regularly distributed neoplastic cells with pleomorphic and hyperchromatic nuclei and moderate amount of cytoplasm. Few studies have documented that tumours with mitotic activity less than 5/10 High Power Field (HPF) showed better survival rate as compared to those with more than 5/10 HPF. Follicle formation of varying sizes and shapes were observed. Call-Exner bodies are noted infrequently as compared to the adult type [11]. The positive IHC stain for Inhibin, an ovarian glycoprotein is an important diagnostic feature along with Calretinin, CD 99 and FOXL2. These markers help in the prognostication of this rare tumour.
Two theories have been proposed to explain the aetiology of sex cord–stromal tumours. The first suggests that GCT in general is derived from the mesenchyme of the developing genital ridge. The second states that sex cord and stromal cells of the mature ovary are derived from precursors found within the mesonephric and coelomic epithelium. Many cell signaling pathways, such as TGF-β, Notch and PI3K/AKT, are involved in the development of GCT. These signal pathways do not act singly but rather form a complex network leading to the formation and development of GCT. Forkhead Box L2 (FOXL2) is one of the most important mutant gene involved in the TGF-β pathway. Mutation of FOXL2 inhibits the activation of SMAD3 by interacting with Bone Morphogenic Proteins (BMP), follistatin and activin A [20]. An interaction between notch signaling and PI3K/AKT has also been proved. Vascular Endothelial Growth Factor (VEGF) and anti-Müllerian hormone (AMH) have also been implicated [19]. Usually, JGCT can be diagnosed as early as stage IA as in present study. Management depends on the age of the patient and dissemination of the disease. There has been no set standard protocol for adjuvant chemotherapy because of the rarity of the disease. Surgery is the primary standard initial treatment. A worse prognosis and early recurrence should be expected in the case of an advanced disease [1]. Unilateral salpingo-oophorectomy is the treatment of choice for children and females in the reproductive age with stage IA disease. Lymph node metastasis is rarely observed in patients with sex-cord stromal tumours, pelvic and para-aortic lymphadenectomy may not be included in the staging surgery of these patients.
The management of sex cord ovarian tumours is limited by their low incidence, wide range of histological pattern and variable biologic behaviour regarding the tumour markers [21]. Inhibin is a peptide hormone produced by ovarian granulosa cells that has a regulatory effect on FSH secretion. The increased levels of Inhibin may return back to the baseline levels after surgery [22]. In one of the patient it was noticed to have advanced bone age and accelerated height velocity presenting with gait disturbances. Consultation with orthopaedician was sought for gait training and high levels of tumour-derived serum estradiol were noted [17].
Generally, the prognosis of JGCT is favourable. Surgical stage is the single most important prognostic factor. The prognosis is poor in the advanced stage than in the adult type of GCT. Adjuvant chemotherapy is recommended in advanced stages [1,9,23]. Recurrence rate is the most crucial factor for mortality in GCT. Nearly, one-fifth of patients with stage I disease recurred after 20 years from diagnosis, thus emphasising the need of long-term follow-up. Targeted therapy for these signaling pathways can eventually prevent recurrence of GCT thus significantly improving the patient survival and also, decrease the resistance of the chemotherapeutic drugs [24,25]. Of all the clinical parameters, tumour stage (FIGO stages) is of greatest prognostic value and must be carefully evaluated in all the patients. Both juvenile- and adult-type GCTs are almost always unilateral [21]. According to the unilateral origin of the tumour, eight cases (6 in stage Ia and 2 cases in stage Ic) were considered as stage I. Unilateral salpingo-oophorectomy was done for stage I [10,25]. After starting of treatment the breast size regressed and serum Estradiol and Inhibin levels started falling.
The study describes the unique features of JGCT. Knowledge of these findings helps in the accurate diagnosis thus classifying them. Since the prognosis of both the subtypes is different so a diagnosis is mandatory. This study also highlights the importance of applying IHC markers for characterisation.
Limitation(s)
The present study was limited due to lesser number of cases. Since, it is a rare entity so even in tertiary level teaching institutions the number of cases was limited.
Conclusion(s)
Although Juvenile Granulosa Cell Tumour occurs frequently during childhood, it is also seen in adults. Primary management of this disease is surgery. The benefit of adjuvant therapy is unclear as survival is long in the early stage and most of the patients are young, fertility sparing surgery can be safely chosen in these patients.