An Autopsy Case Report of Von Hippel-Lindau Syndrome-Type 2C Masquerading as Pre-eclampsia and Resulting in Maternal and Foetal Mortality
Gwendolyn Fernandes1, Varsha Deshpande2
1 Additional Professor, Department of Pathology, G.S Medical College and K.E.M. Hospital, Mumbai, Maharashtra, India.
2 Fellow in Uropathology, Department of Pathology, G.S Medical College and K.E.M. Hospital, Mumbai, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Gwendolyn Fernandes, C-802, Swayam, Poonam Gardens, Mira Road, Thane District, Mumbai, Maharashtra, India.
E-mail: drgwenfern@yahoo.co.in
A 23-year-old female with seven months pregnancy, presented with gestational hypertension associated with mild proteinuria and was diagnosed clinically as pre-eclampsia and referred to tertiary care centre. She also had progressive breathlessness and giddiness. On examination, pallor, Blood pressure (BP) of 180/100 mm Hg and crepitations in left lower lung were noted. Laboratory investigations revealed neutrophilic leucocytosis and a profile of Disseminated Intravascular Coagulation (DIC). An ultrasound (USG) of the abdomen showed a gravid uterus with Intrauterine Foetal Death (IUFD) and a left adrenal mass. The clinical diagnosis was acute hypertensive heart failure with severe pre-eclampsia. She was put on three anti-hypertensives and ventilatory support, but she succumbed to her illness, within three days of admission. A complete autopsy was performed. The autopsy revealed hypertensive heart disease with marked left ventricular hypertrophy, severe coronary atherosclerosis, cystic bilateral adrenal pheochromocytoma, a cystic para-aortic paraganglioma, a pancreatic Neuroendocrine Tumour (NET) with lymph node metastasis. Hence, a diagnosis of DIC with refractory hypertension in Type 2C Von Hippel-Lindau (VHL) syndrome was established. VHL syndrome is an autosomal dominant disorder caused by germline VHL mutation which leads to development of various tumours. VHL disease is classified into four subtypes and VHL type 2C has predominantly pheochromocytomas without haemangioblastoma and Renal Cell Carcinoma (RCC). As the patient mimicked pre-eclampsia clinically, the diagnosis of VHL was missed. Bilateral adrenal cystic masses with gestational hypertension should be thoroughly evaluated, at the earliest, for familial pheochromocytomas or VHL syndromes as they may prove fatal.
Gestational hypertension, Pheochromocytoma, Refractory hypertension
Case Report
A 23-year-old female, gravid 2, para 0, living 0, intra uterine fetal death 2, was referred to the tertiary care centre with seven months pregnancy complicated by severe pre-eclampsia, refractory hypertension and IUFD. She also had giddiness and weakness since one month and gradual worsening of breathlessness from grade I to grade IV, since 2-3 days. There was no history of pain in abdomen, blurring of vision, convulsions or loss of consciousness. She had a history of chronic hypertension with a BP of 180/100 mm Hg which resulted in a third trimester IUFD. The patient did not take any anti-hypertensives after the pregnancy despite medical advice. There was no family history of hypertension or any other disease.
On admission, her BP was 220/110 mm Hg for which two doses of Inj Labetalol was given, after which her BP decreased but remained at 190/110 mm Hg. Pallor as well as crepitations in the left lower lung were noted.
Laboratory investigations revealed the following: Haemoglobin was 8.8 gm/dL; Total leucocyte count was 24.8×109/L with 87% neutrophils, 10% lymphocytes and 3% eosinophils. Platelet count was 2.24×109/L. Peripheral blood smear showed a hypochromic microcytic picture and adequate platelets. Her coagulation profile was deranged and showed raised D-Dimer (8.9) (Ref range: 0-0.49), raised fibrin degradation products (17.26) (Ref range: <3.79). Fibrinogen was within normal limits (389 mg/dL) (Ref range: 3rd trimester-280-590). Prothrombin time was 13.5 seconds and activated partial thromboplastin time was 27.2 seconds. Urine examination showed albumin levels of 30 mg/dL, with 12-15 pus cells/hpf and 6-8 epithelial cells/hpf. Urinary glucose was absent. USG abdomen showed a single intra-uterine pregnancy of 26 weeks with absent cardiac activity. A large suprarenal cystic mass measuring 12×10 cm was also seen. Electrocardiography and 2D Echocardiography showed left ventricular hypertrophy. Rest of the investigations were within normal limits.
In view of the above, a clinical diagnosis of severe pre-eclamsia with acute hypertensive heart failure was made. She was treated with tablet. Metoprolol 50 mg stat followed by Inj NTG in 100cc NS @15 mL/hr. She was also given Inj Lasix 40 mL IV stat, Inj Fentanyl 12.5 mL/hr, Inj Labetalol 20 mg IV SOS, Inj DOPA 400 mg in 50 mL NG @15 mL/hr., tablet. Nicardia R (30) QID and tablet. Prazopressin 2.5 mg. In spite of all the treatment, her BP continued to remain at 190/110 mm of Hg and her general condition kept worsening. Terminally, she was mentally disoriented and had tachypnoea. She was given cardio pulmonary resuscitation and put on mechanical ventillation. Inj Atropin 2cc and Inj Adrenalin 2cc were given. She succumbed to the illness and a complete autopsy was performed.
At postmortem examination, pallor was present but no cyanosis, icterus and clubbing was seen. Heart was moderately enlarged in size. Left anterior descending artery showed critical stenosis by an eccentric atheromatous plaque for 2 cm. Prominent left ventricular hypertrophy and uncomplicated atherosclerosis of the entire thoraco-abdominal aorta was seen.
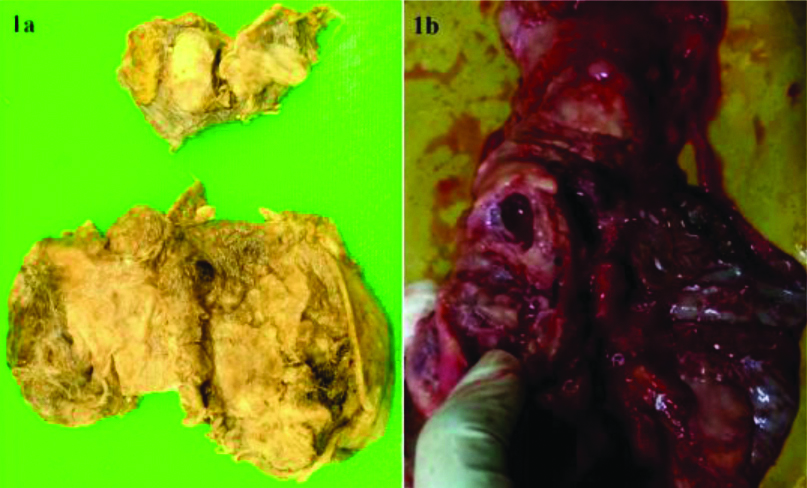
A para-aortic cystic mass measuring 2.5 cm was found on the outer aspect of abdominal aorta. Both adrenal glands were enlarged in size with solid-cystic morphology. Solid-cystic masses of the left and right adrenal gland, measuring 12×10 cm and 3×2 cm, respectively were seen [Table/Fig-1a]. The pancreas showed a creamy coloured cystic mass measuring 1 cm in diameter [Table/Fig-1b] with enlarged peri-pancreatic lymph nodes and an enlarged porta hepatis lymph node measuring 2 cm. The remaining organs were unremarkable.
a) Bilateral pheochromocytoma (Left- 12×10 cm, Right- 3×2 cm); b) Pancreas showing a creamy-coloured cystic Neuroendocrine Tumour (NET) measuring 1 cm in diameter.
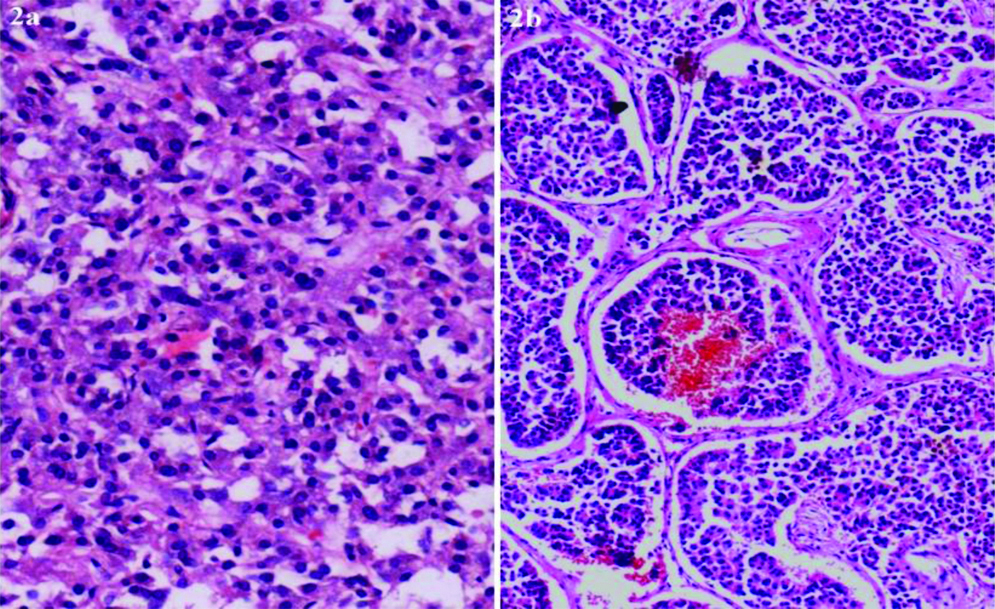
On histopathology, both adrenal masses showed classic morphology of a pheochromocytoma with Zellballen pattern composed of small nests of polygonal eosinophilic cells surrounded by a fibro-vascular stroma [Table/Fig-2a]. The para-aortic paraganglioma also showed similar histological features. The pancreas showed a well differentiated Neuroendocrine Tumour (NET), composed nests of small polygonal cells with stippled nuclear chromatin [Table/Fig-2b]. Metastasis of the pancreatic NET to peripancreatic and porta hepatis lymph nodes was seen. (<1 mitotic figure/10 HPF and MIB-1 index of <1% seen) Immunohistochemistry with synaptophysin and chromogranin showed strong positivity and confirmed NET of the pancreas and pheochromocytoma. No haemangioblastoma or RCC were seen. The final cause of death certified at autopsy was DIC in a case of refractory hypertension due to type 2C VHL with IUFD. Genetic analysis could not be done for economic reasons. Genetic analysis was not required for the diagnosis but would help in counselling of the family members.
a) Pheochromocytoma showing polygonal cell nests separated by fibrovascular stroma (H&E x 400). b) Pancreatic Neuroendocrine Tumour (NET) showing organoid arrangement of tumour cells (H&E, x 200).
Discussion
Von Hippel-Lindau (VHL) is a rare autosomal dominant disorder caused by germline mutations in the VHL tumour suppressor gene that results in the occurrence of multiple endocrine and non-endocrine lesions. The spectrum of tumours includes haemangioblastomas, RCC, pheochromocytomas and paragangliomas, pancreatic serous cystadenomas and pancreatic NET and endolymphatic sac tumours [1-3].
The incidence of VHL is 1 in 36,000 live births. 80% cases are familial while, 20% patients will present with de novo mutations. VHL has >90% penetrance in patients over 65 years of age [1]. Carriership of a germline mutation in the VHL gene predisposes to the VHL-disease. VHL gene is a tumour suppressor gene and a tumour cell develops when inactivation of both copies of the VHL gene occurs. Specific tumour mutations may be associated with different tumour susceptibility risks [4]. VHL disease shows variable expression and interfamilial differences in the predisposition to phaeochromocytoma [4].
VHL is classified into 4 subtypes: type 1, type 2A, type 2B and type 2C. VHL type 1 is characterised by retinal as well as CNS haemangioblastomas and RCC, without pheochromocytoma. Type 2A is characterised by haemangioblastoma and RCC while type 2B has haemangioblastomas, renal carcinoma and pheochromocytomas [1]. Type 2C has been recently identified as a separate group and is characterised exclusively by pheochromocytomas without haemangioblastomas and RCCs, as seen in our case. Our case showed bilateral pheochromocytomas, a para-aortic paraganglioma and in an addition, a metastatic pancreatic NET.
Pheochromocytomas have been noted in 30% of VHL patient series. They may present with headache, palpitations, tremors, diaphoresis and severe hypertension. In the present case, abundant catecholamine production by the pheochromocytomas and paraganglioma led to refractory hypertension with her BP persisting at 190/110 mm Hg, despite aggressive treatment [5].
Pancreatic NETs are observed in 11-17% of patients with VHL. VHL patients with pancreatic NETs are slow growing, seen in younger patients and are non-functional as was seen in present case. The median Ki-67 proliferation index is low at 3% and lymph node metastasis was seen in 43% of the cases reported in literature [1]. The pancreatic NET in present case, had a MIB-1 proliferation index of <1% and was a grade I NET but showed metastasis to peripancreatic and porta hepatic lymph nodes. Metastasis would have determined the outcome, if patient had survived the hypertensive emergency.
A clinical diagnosis of pre-eclampsia was made, as in the present case patient presented with refractory hypertension as well as mild proteinuria. She also had chronic hypertension diagnosed in previous pregnancy and the hypertension persisted in the current pregnancy. Bilateral pheochromocytomas and paraganglioma which caused the refractory hypertension were diagnosed only at autopsy. High catecholamine levels were the cause of the refractory hypertension, and resulted in IUFDs in both the pregnancies. VHL in this case masqueraded as pre-eclampsia and unfortunately resulted in mortality of both mother and child. Majority of pregnant women with VHL have a favorable pregnancy outcome with low maternal mortality and high foetal survival, if diagnosed in time [6]. As VHL is a complex disease and multiple organs may be involved at different times in the life of the patient, an interdisciplinary approach in treatment is needed [7].
Conclusion(s)
Patients with refractory hypertension or past history of pregnancy induced hypertension with adrenal masses should be thoroughly evaluated, at the earliest, for familial pheochromocytomas or VHL syndromes, as they may prove fatal.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Apr 09, 2020
Manual Googling: May 25, 2020
iThenticate Software: Jun 13, 2020 (13%)
[1]. Lloyd RV, Osamura RY, Kloppel G, Rosai J, (2017), WHO Classification of Tumours of Endocrine organs 2017 4th editionLyonIARC:257-61. [Google Scholar]
[2]. Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Clinical features and natural history of von Hippel-Lindau diseaseQ J Med 1990 77(283):1151-63.10.1093/qjmed/77.2.11512274658 [Google Scholar] [CrossRef] [PubMed]
[3]. Maher ER, Neumann HP, Richard S, von Hippel-Lindau: A clinical and scientific reviewEur J Hum Genet 2011 19(6):617-23.10.1038/ejhg.2010.17521386872 [Google Scholar] [CrossRef] [PubMed]
[4]. Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, Phenotypic expression in Von Hippel-Lindau disease: Correlations with germline VHL gene mutationsJ Med Genet 1996 33(4):329-32.10.1136/jmg.33.4.3288730290 [Google Scholar] [CrossRef] [PubMed]
[5]. Zuber SM, Kantorovich V, Pacak K, Hypertension in pheochromocytoma: characteristics and treatmentEndocrinol Metab Clin North Am 2011 40(2):295-311.10.1016/j.ecl.2011.02.00221565668 [Google Scholar] [CrossRef] [PubMed]
[6]. Merhi B, Miller M, Lanis A, Katz B, Hsu T, Tong I, Management of uncommon disorders in pregnancy: Von Hippel-Lindau disease, Gitelman syndrome, and Nutcracker syndromeObstet Med 2017 10(3):138-41.10.1177/1753495X1668308829051782 [Google Scholar] [CrossRef] [PubMed]
[7]. Schmid S, Gillessen S, Binet I, Brandle M, Engeler D, Greiner J, Management of Von Hippel-Lindau disease: An interdisciplinary reviewOncol Res Treat 2014 37(12):761-71.10.1159/00036936225531723 [Google Scholar] [CrossRef] [PubMed]