Collagenofibrotic Glomerulopathy: A Rare Diagnosis and Seldom Thought of Differential for Nodular Glomerular Mesangial Expansion
Anisha Manocha1, Pallav Gupta2
1 Senior Resident, Department of Pathology, Sir Ganga Ram Hospital, New Delhi, India.
2 Consultant, Department of Pathology, Sir Ganga Ram Hospital, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Pallav Gupta, Department of Pathology, Sir Ganga Ram Hospital, Old Rajender Nagar, New Delhi, India.
E-mail: pallavkmc1@gmail.com
Collagenofibrotic glomerulopathy, a very rare glomerular disease, characterised by abnormal deposition of Type III collagen in the mesangium and subendothelial space. This disease may have an indolent course clinically but is capable of finally progressing to end stage renal failure. Thus, it is imperative for clinicians and pathologists to be mindful of this entity due to its rarity, nonspecific presenting signs and symptoms and varied differentials on histopathology which must be ruled out. A definite diagnosis depends on the awareness of this entity, a high index of suspicion and confirmation by immunohistochemistry and electron microscopy. Here, the present authors report a case of a 41-year-old hypertensive female who came with persistent pedal oedema and proteinuria. The biopsy showed nodular expansion of the mesangium by a homogenous, Congo red negative, eosinophilic material. A diagnosis of Collagenofibrotic glomerulopathy was confirmed on immunohistochemistry and electron microscopy.
Collagen Type III, Glomerular mesangium, Kidney injury, Nephrotic syndrome, Proteinuria
Case Report
A 41-year-old hypertensive, non-diabetic female presented with chief complaint of persistent pedal oedema since four months. Her systemic examination was normal. Urinalysis revealed 3+ proteinuria. Serum creatinine level was 1.17 mg/dL (0.7-1.25mg/dl) and serum potassium was 3.5 mEq/L (3.5-5.10mEq/L). The routine urine microscopy revealed inactive sediment, 8-10 pus cells and 8-10 RBCs. Urine protein electrophoresis was normal. Ultrasound showed a normal sized kidney.
She had presented with similar complaints earlier and was found to have had an episode of nephrotic syndrome four years ago. She was evaluated for proteinuria and pedal oedema elsewhere. A renal biopsy performed at that time was reported as focal segmental glomerulosclerosis. Initially, she was given steroids for a year but subsequently developed resistance and was switched to Rituximab. Following this, a combination of Tacrolimus and Wysolone was given.
The renal biopsy was repeated at the present institute and showed 14 glomeruli, all of which showed nodular mesangial expansion by an acellular eosinophilic material which was negative for Periodic acid Schiff (PAS) and Periodic acid Silver Methenamine (PSM) [Table/Fig-1a]. Some of the tubules showed hyaline casts. The interstitium and blood vessels were unremarkable. In view of a previous episode of nephrotic syndrome, current nephrotic range proteinuria and nodular mesangial expansion by an acellular eosinophilic material, a histologic differential diagnosis of amyloidosis was considered first. However, the Congo red stain was negative.
a) Nodular expansion of the mesangium (white arrow) by a homogenous eosinophilic material (X200; H&E); b) Positive staining (white arrow) for Collagen Type III on immunohistochemistry (X100, IHC); c) Expansion of the subendothelial and mesangial space (arrow mark) by a banded fibrillary material on electron microscopy (X2000; uranyl acetate and lead citrate); d) Fibrillary material with banded appearance and a periodicity of 30-60 nm on electron microscopy (X10000; uranyl acetate and lead citrate).
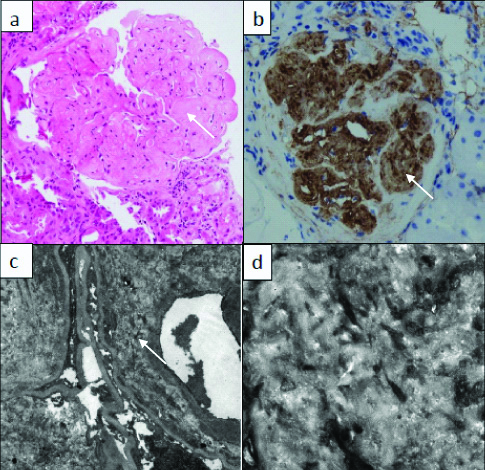
The other differentials considered were diabetic glomerulopathy and light chain deposition disease. However, absence of any history of diabetes and lack of PAS positivity in the mesangial nodules ruled out diabetes while direct immunofluorescence was performed to rule light chain deposition disease. Direct immunofluorescence showed negativity for all the immunoglobulins, complements and light chains. Therefore, light chain deposition disease or any other immune complex deposition disease was excluded. In view of nodular mesangial expansion and Congo red, PAS and PSM negativity, a diagnosis of Collagenofibrotic glomerulopathy was suggested after ruling out all other differential diagnosis. Electron microscopy and Immunohistochemistry for Type III collagen was advised.
Immunohistochemistry was positive for Type III collagen within the glomerulus [Table/Fig-1b]. Electron microscopy showed mesangial and subendothelial space expansion by a banded fibrillary material [Table/Fig-1c,d]. Thus, Collagenofibrotic glomerulopathy was confirmed as the final diagnosis. The patient was given supportive treatment along with albumin infusion and diuretics and was discharged in a stable condition on Wysolone 20 mg and Endoxan 100 mg once daily. This was followed for a period of 2 months after which the dosage of steroids was tapered. After five months of follow-up, patient was stable without any pedal oedema.
Discussion
Collagenofibrotic glomerulopathy is an extremely rare glomerular disease characterised by abnormal deposition of Type III collagen in mesangium and subendothelial space. It is also accompanied by a marked increase in the serum Type III pro-collagen peptide levels [1]. Normally, Type III collagen is present on the interstitium and blood vessels, but not in the glomeruli. The glomeruli also normally express Type IV, V and VI collagen, but not Type III. However, dysregulation of Type III collagen results in its’ deposition in the mesangium, basement membrane or subendothelium and promotes fibrosis. This dysregulation of Type III collagen results in two diseases- Collagenofibrotic glomerulopathy and Nail patella syndrome [2]. Initially, Collagenofibrotic glomerulopathy was considered to be a variant of nail-patella syndrome with no skeletal deformities [1]. First described by Arakawa M, it was subsequently included in the WHO classification of glomerular diseases since 1995 [3,4]. Collagen Type III glomerulopathy and primary glomerular fibrosis are other names documented in literature for this entity [5,6].
Owing to its infrequency, Collagenofibrotic glomerulopathy has had a sluggish progress in the understanding of its pathogenesis and treatment. There are fewer than 40 cases documented in literature for this entity. A significant number have been reported from Japan and Asian countries and about 17 are from India [7]. The largest series has been reported by Kurien AA et al., from India and Gubler MC et al., from France comprising of 8 adult cases and 10 paediatric cases respectively [7, 8]. Although no definite age or gender predilection has been documented in literature, maximum cases were from the fourth to seventh decade. While this disease is more common in adults, cases in children have also been reported and children are more likely to progress to end stage renal disease [9].
It is crucial for pathologists and clinicians to be aware of this entity because the diagnosis depends heavily on a high index of suspicion. This is because this disease does not present with any characteristic signs and symptoms, but in due time can progress to end stage renal disease. Furthermore, varied differential diagnosis on histopathology makes a definite diagnosis all the more difficult. Clinically, collagenofibrotic glomerulopathy most commonly presents with nephrotic range proteinuria and microscopic haematuria, accompanied by other nonspecific symptoms such as oedema and hypertension with minor alterations in the renal function. This was similar to the present case, where patient chiefly presented with persistent proteinuria. However ultimately, it may result in end stage kidney disease [7]. As the symptoms are nonspecific it is often not on the clinical list of differentials at the time of biopsy. It usually presents as a sporadic isolated form in adults, and may rarely also present as a familial autosomal recessive form in children [8].
The aetiopathogenesis is still unclear. Some authors believe it to be a systemic disorder with abnormal metabolism of Type III collagen whereas others have suggested a localised involvement where abnormal Type III collagen is produced endogenously by the mesangium. The role of interleukin-4 has also been described, as it selectively stimulates Type III collagen synthesis in the glomerulus. Due to the clustering of cases from Japan and Asia, a role of genetic factors has also been suspected [9].
On light microscopy our main differentials were- Amyloidosis, Diabetic glomerulosclerosis and light chain deposition disease. The present case lacked Congo red positivity of amyloidosis and history and PAS positivity of diabetic glomerulopathy. In light chain deposition disease, mesangial nodules are more in number and uniform in distribution. They also show either kappa or lambda positivity on immunofluorescence.
Furthermore, immunofluorescence in this case showed negativity for all immunoglobulins, complements and light chains. Banded fibrillary collagen in mesangium and subendothelial space on electron microscopy as well as positivity for Type III collagen on immunohistochemistry is mandatory for diagnosis.
Collagenofibrotic glomerulopathy can be distinguished from nail patella syndrome only on electron microscopy, by the relative sparing of lamina densa of the glomerular basement membrane which is involved in the latter. Collagenofibrotic glomerulopathy also lacks the LMX1B gene mutations identified in the inherited disorder of Nail patella syndrome [10].
No specific treatment protocol has been documented for this condition. Symptomatic treatment such as control of hypertension and oedema may help. Dialysis or renal transplant may be suggested in end stage renal disease. No recurrences have been documented so far in literature [11].
Conclusion(s)
Collagenofibrotic glomerulopathy is a very rare disease which requires a high index of suspicion and awareness of this entity by the pathologist. It has varied differentials on histopathology which must be ruled out before this diagnosis is made. Immunohistochemistry and electron microscopy are crucial to identify deposits of Type III collagen. Also, any deposition disease must be examined by electron microscopy for exact categorisation and confirmation of the diagnosis.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Mar 03, 2020
Manual Googling: Apr 24, 2020
iThenticate Software: May 11, 2020 (5%)
[1]. Cohen AH, Collagen type III glomerulopathiesAdv Chronic Kidney Dis 2012 19(2):101-06.10.1053/j.ackd.2012.02.01722449347 [Google Scholar] [CrossRef] [PubMed]
[2]. Anitha A, Vankalakunti M, Siddini V, Babu K, Bonu R, Ballal S, Type III collagen disorders: A case report and review of literatureIndian J Pathol and Microbiol 2016 59(1):75-77.10.4103/0377-4929.17482226960642 [Google Scholar] [CrossRef] [PubMed]
[3]. Arakawa M, Idiopathic mesangio-degenerative glomerulopathyJpn J Nephrol 1979 21:914-15. [Google Scholar]
[4]. Churg J, Bernstein J, Glassock RJ, Classification and atlas of glomerular diseases 1995 2nd ednIgaku-Shoin, New York, NY [Google Scholar]
[5]. Ikeda K, Yokoyama H, Tomosugi N, Kida H, Ooshima A, Kobayashi K, Primary glomerular fibrosis: A new nephropathy caused by diffuse intra-glomerular increase in atypical type III collagen fibersClin Nephrol 1990 33(4):155-59. [Google Scholar]
[6]. Imbasciati E, Gherardi G, Morozumi K, Gudat F, Epper R, Basler V, Collagen type III glomerulopathy: A new idiopathic glomerular diseaseAm J Nephrol 1991 11(5):422-29.10.1159/0001683501809042 [Google Scholar] [CrossRef] [PubMed]
[7]. Kurien AA, Larsen CP, Cossey LN, Collagenofibrotic glomerulopathyClin Kidney J 2015 8(5):543-47.10.1093/ckj/sfv06126413279 [Google Scholar] [CrossRef] [PubMed]
[8]. Gubler MC, Dommergues JP, Foulard M, Bensman A, Leroy JP, Broyer M, Collagen type III glomerulopathy: A new type of hereditary nephropathyPediatr Nephrol 1993 7(4):354-60.10.1007/BF008575368398640 [Google Scholar] [CrossRef] [PubMed]
[9]. Duggal R, Nada R, Rayat CS, Rane SU, Sakhuja V, Joshi K, Collagenofibrotic glomerulopathy- A reviewClin Kidney J 2012 5(1):07-12.10.1093/ndtplus/sfr14426069739 [Google Scholar] [CrossRef] [PubMed]
[10]. Matthai SM, Mohapatra A, Duhli N, David VG, Varughese S, Collagenofibrotic glomerulopathy- A rare disease diagnosed with the aid of transmission electron microscopyIndian J Pathol Microbiol 2020 63(5):47-49.10.4103/IJPM.IJPM_341_1832108627 [Google Scholar] [CrossRef] [PubMed]
[11]. Patro KC, Jha R, Sahay M, Swarnalatha G, Collagenofibrotic glomerulopathy-Case report with review of literatureIndian J Nephrol 2011 21(1):52-55.10.4103/0971-4065.7808021655172 [Google Scholar] [CrossRef] [PubMed]