Epidermolysis Bullosa in Newborn: A Rare Case with Management Dilemmas
Ekta Kale1, Sumita Mehta2, Tarun Kumar3
1 Medical Officer, Department of Obstetrics and Gynaecology, Babu Jagjivan Ram Memorial Hospital, Delhi, India.
2 Specialist, Department of Obstetrics and Gynaecology, Babu Jagjivan Ram Memorial Hospital, Delhi, India.
3 Specialist, Department of Paediatrics, Babu Jagjivan Ram Memorial Hospital, Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sumita Mehta, Babu Jagjivan Ram Memorial Hospital, Delhi, India.
E-mail: sumitadr@gmail.com
Epidermolysis Bullosa (EB) is a rare genetic and connective tissue disorder affecting 1 in every 50000 live birth that causes skin to be very fragile and blister easily. Epidermolysis Bullosa Simplex (EBS) is the most common of all EB and it presents at birth or during the neonatal period. Diagnosis of EB is mainly clinical and can be confirmed by genetic analysis of the parents and the patient. There is presently no definite cure for EB and treatment is only conservative to alleviate symptoms and provide supportive care. We present a case report of a neonate born with reddish discolouration and peeling of skin of fingers of both hands which progressively developed into fluid filled vesicles over the abdomen and buttocks. The biopsy of the lesions was deferred due to financial constraints and a clinical diagnosis of EBS was made. The newborn was started on injectable antibiotics and given supportive therapy in the form of daily wound care and protective bandaging with which she improved and was discharged on 10th postnatal day in stable condition.
Butterfly children, Epidermolysis bullosa simplex, Skin blisters, Skin fragility
Case Report
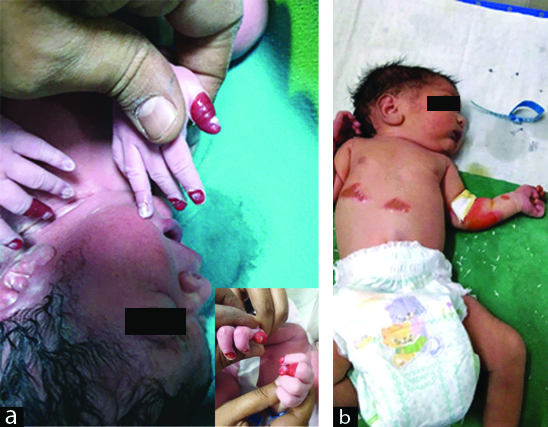
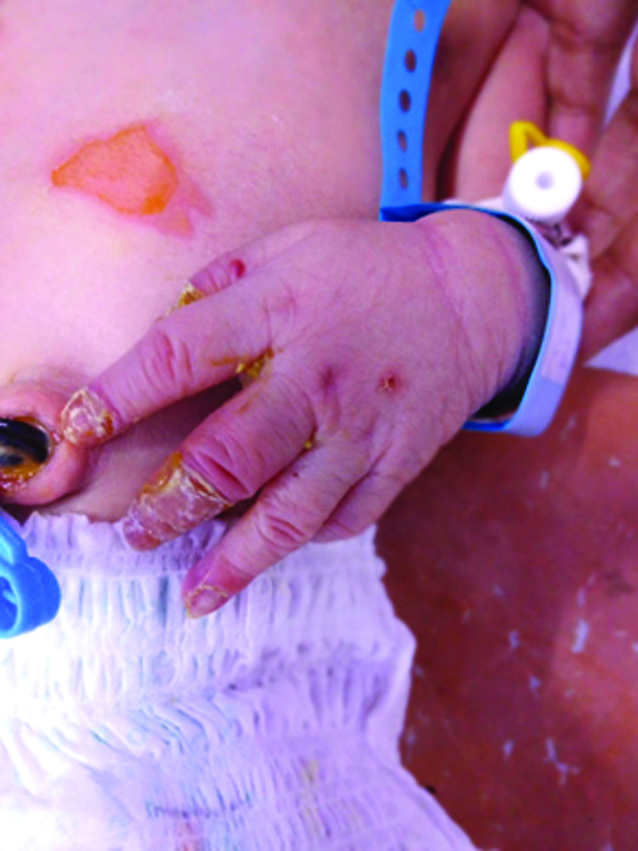
A 26-year-old primigravida booked patient was admitted at 36 weeks 4 days of gestation with oligohydroamnios. Her antenatal period was uneventful and all her blood investigations were normal; USG colour Doppler showed a single live foetus of 35 weeks + 3 days with AFI of 5 and low resistance flows in MCA. She was induced with two doses of PgE2 gel after completing a course of inj. Dexamethasone for foetal lung maturity. She went into labour and emergency LSCS was done for foetal bradycardia. A live female baby of birth weight 2.5 kg was delivered with an Apgar score of 8, 9, and 9 at 1, 5 and 10 minutes, respectively. The neonate at birth was noticed to have reddish discolouration and peeling of skin of fingers of both hands. On second postnatal day, she developed blisters and subsequent peeling of skin from the elbows and abdomen. These were the areas where minor trauma had occurred in the form of attachment of adhesive tape (micropore) and abdominal temperature probe [Table/Fig-1a,b]. The neonate also developed white plaques in the oral cavity which were adherent to the palate and upper labial mucosa. Her ophthalmic examination was normal. Rest of systemic examination was normal and the neonate did not have any feeding or respiratory difficulty. Biopsy of the lesions was deferred due to financial constraints and a clinical diagnosis of EBS was made after consultation with the dermatologist. Baby was managed conservatively and nursed on soft paraffin gauze along with mupirocin ointment dressing to prevent trauma. She was started on antibiotics (Inj Cefotaxime and Inj vanconycin) and daily wound care and preventive bandaging was done. By 7th postnatal day the lesions had healed to a large extent [Table/Fig-2]. Baby was discharged on postnatal day 10 in stable condition with parents counselled regarding gentle handling of the child and care for the lesions.
(a): Spontaneous peeling of skin of fingers of both hands at birth. (b) Blister formation and peeling of skin on elbow and trunk.
Areas of peeling skin undergoing healing.
Discussion
Epidermolysis bullosa is an autosomal dominant disorder seen in 1 in 50,000 live births [1]. The skin of the affected neonate is very fragile and has been compared to the delicate wings of a butterfly and such children are often referred as “butterfly children” [2]. The third International Consensus meeting on diagnosis and classification of EB held in 2008 categorised EB into more than 30 subtypes based on their phenotypic characteristics, gene involved, mutation and immunohistochemistry [3]. Based on the distribution of blisters, EB is divided into 4 types- EB simplex, junctional EB, dystrophic EB and Kindler syndrome. EBS is the most common and milder variant and results from mutations in KRT5 or KRT14 genes which are responsible for keratin 5 and 14 protein synthesis. As a result of deficiency of these proteins, the epidermis becomes fragile and is easily damaged even in response to minor trauma or friction. The neonate in present case was also born with skin peeling of fingers of both hands and her skin was so fragile that it underwent blister formation and subsequent peeling with such minor trauma as application of adhesive tape and temperature probe on second postnatal day. Other cutaneous features including keratoderma, Koebner’s blisters (commonly seen on scalp, hands and feet) and dyspigmentation have been reported by various authors [2,4]. In addition, oral and ocular mucous membranes can also be involved. In present case also, the oral cavity had presence of white plaques adherent to the palate in addition to the lesions present on the limbs and trunk.
The diagnosis of EB is mainly based on positive family history and clinical presentation. The superficial peeling of skin without bleeding strongly points towards EB which can be further confirmed by genetic analysis of the newborn and the parents. Further, to correctly identify the proteins affected and the plane of separation, a skin biopsy and immunofluorence mapping is essential. Diagnosis in present case was also made clinically with the pathognomonic blister formation and peeling of fragile skin in response to minor trauma. Facility for genetic analysis was not available with us and was omitted. Blisters of EBS usually heal without leaving scars. Secondary infection is the most dreaded complication and severe cases with widespread blisters, can have infection, dehydration and other medical problems which can even be life threatening to the infant. There is presently no definite cure for EB and objective of treatment is to alleviate symptoms and provide supportive care. Key to successful management in these cases is expert nursing care and special precautions should be taken to protect the baby from undue trauma induced blisters which can be caused by use of adhesive tapes, tourniquets, BP cuffs. Areas prone to develop blisters should be protected by padding and open wounds should be dressed with non-adherent dressing pads or Vaseline impregnated guaze. Blisters have a propensity to enlarge and so they are punctured with a sterile needle keeping the roof intact to prevent subsequent infection [5,6]. In present case also, baby was managed conservatively and nursed on thick soft foam pad, antibiotics were started to prevent infection and anti-fungal mouth paint was used to maintain oral hygiene. Counselling the parents regarding care of the newborn with special emphasis on prevention of trauma and gentle handling is of utmost importance. Different evidence based therapies based on individual mutations, disease mechanisms and phenotypic considerations are being developed for severe forms of EB [7,8]. Genetic counseling should be offered to prospective parents who have a family history of EB and Chorionic Villus Sampling (CVS) can be done at 8-10 weeks of pregnancy in couples at high risk of having a child with EB.
Conclusion(s)
Epidermolysis bullosa is a rare group of genetic disorders that causes the skin to be very fragile and causes blister easily. The diagnosis is mainly clinical and is confirmed by genetic analysis of the patient and the parents. There is presently no cure for EB and so genetic counselling to the prospective parents who have a family history of EB or offering CVS to couples at high risk are the only options to decrease incidence of EB. Management of various forms of EB is complex and requires interdisciplinary collaboration. The main objective of treating patients with EB is to alleviate symptoms and provide supportive treatment. Different evidence based approaches including gene therapies, cell and protein therapy are currently being tested at preclinical and clinical levels.
Author Declaration:
Financial or Other Competing Interests: No
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Oct 22, 2019
Manual Googling: Dec 21, 2019
iThenticate Software: Dec 29, 2019 (8%)
[1]. Maldonado-Colin G, Hemandez-Zepeda C, Duran-McKinster C, Garcia-Romero MT, Inherited epidermolysis bullosa: A multisystem disease of skin and mucosae fragilityIndian J Paediatr Dermatol 2017 18:267-63.10.4103/ijpd.IJPD_16_17 [Google Scholar] [CrossRef]
[2]. Reddy RS, Shrikande DY, Nigwekar P, Panwar S, Patil P, Epidermolysis bullosa in newborn: A rare case reportIndian Journal of Basic and Applied Medical Research 2014 3(3):131-34. [Google Scholar]
[3]. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, The classification of inherited EB: Report of the third international consensus meeting on diagnosis and classification of EBJ Am Acad Dermatol 2008 58:931-50.10.1016/j.jaad.2008.02.00418374450 [Google Scholar] [CrossRef] [PubMed]
[4]. Maharaja Parmanantham Sekar Epidermolysis bullosa associated septicaemia in a neonate: Case reportBiomed & Pharmacol J 2016 9(2):533-36.10.13005/bpj/969 [Google Scholar] [CrossRef]
[5]. Gonzalez ME, Evaluation and treatment of the newborn with EBSemin Perinatol 2013 37:32-39.10.1053/j.semperi.2012.11.00423419761 [Google Scholar] [CrossRef] [PubMed]
[6]. Denyer J, Management of severe blistering disordersSemin Neonatol 2000 5:321-24.10.1053/siny.2000.001711032716 [Google Scholar] [CrossRef] [PubMed]
[7]. Gostynski AZ, Pasmooij AM, Jonkman ME, Successful therapeutic transplantation of revertant skin in EBJ Am ACad Dermatol 2014 70:98-101.10.1016/j.jaad.2013.08.05224176523 [Google Scholar] [CrossRef] [PubMed]
[8]. Bruckner-Tuderman L, Newer treatment modalities in epidermolysis bullosaIndian Dermatol Online J 2019 10(3):244-50.10.4103/idoj.IDOJ_287_1831149565 [Google Scholar] [CrossRef] [PubMed]