Turner syndrome is a ‘complex developmental disorder in females caused by complete or partial absence of the second sex chromosome (monosomy X) X or Y, resulting in haploinsufficiency of multiple genes’. It is the only monosomy that is compatible with life. Turner syndrome affects almost 1 in 2,500 live female births. The most common phenotype observed in Turner patients is 45,X in more than 50% followed by isochromosome in about 5-10% subjects. Presence of similar copies of one arm of the chromosome and none of the other is defined as the isochromosome, resulting in partial monosomy of one arm and trisomy of the other which could be attributed to the abnormal transverse misdivision of centromere taking places during the meiotic or postzygotic cell division in the premeiotic gamete. The proband in the case report was referred for short stature and she revealed a rare Turner mosaic mos 45,X[6]/47,X,i(X)(q10),i(X)(q10)[6]/46,X,i(X)(q10)[88] karyotype. This article aimed to delineate the genotypic and phenotypic features exhibited by the patient.
Genotype, Isochromosome, Misdivision, Phenotype
Case Report
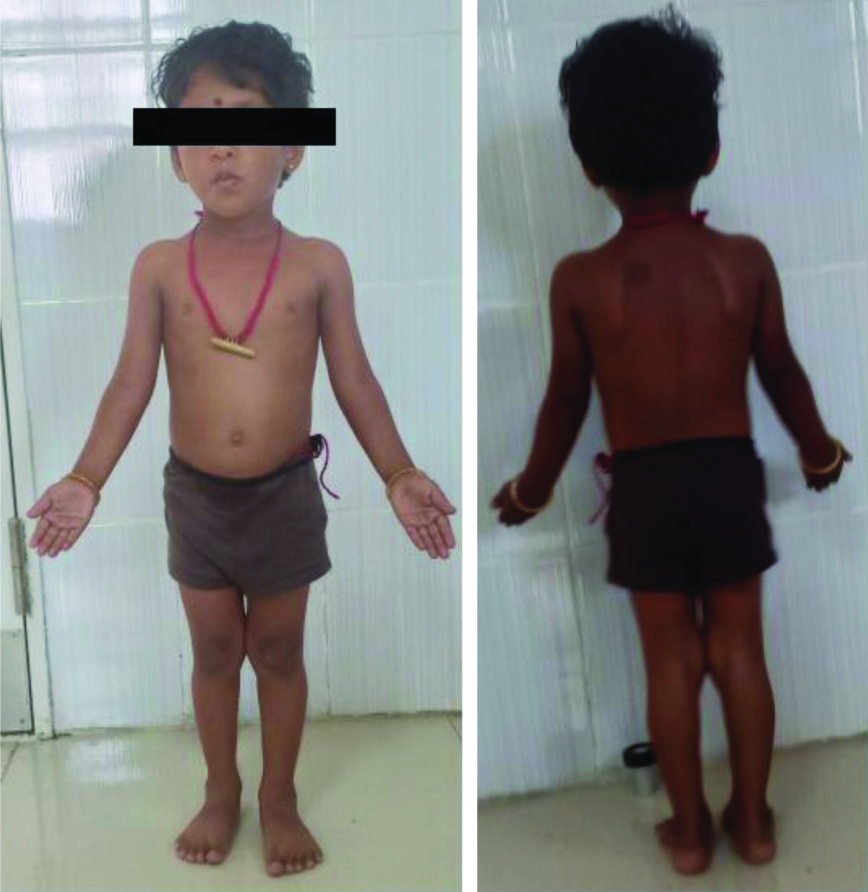
A six-year-old girl child was brought by her parents with chief complaints of short stature (85 cm). She was the third child of healthy non-consanguineous partners. Both of her parents were of below average height, with mid-parental height of 145 cm. Her birth weight was 2.6 kg and her weight at the time of presentation was 11 kg. Based on the anthropometric measurements, both her weight and height were less than 3 percentile. Her developmental stages were normal for age. The proband presented frontal bossing but no other facial dysmorphism. Her skeletal survey tests revealed her bone age to be of 2.6 years. No significant physical abnormality was observed in the patient other than short stature [Table/Fig-1]. Based on the complaints, she presented she was categorised under the provisional diagnosis of Turner Syndrome (TS)/Growth Hormone Deficiency (GHD). She was suggested to undergo thyroid, gonadotropin, growth hormone and ultrasonography study.
Clinical picture of the mosaic isochromosome patient showing no features of Turner syndrome.
Her results indicated that she had normal thyroid functions but her FSH and serum cortisol values were high for her age and the estradiol levels were also low [Table/Fig-2]. Her growth hormone values after the stimulation test were 0.92 ng/mL, 1.3 ng/mL, 28 ng/mL and 10.2 ng/mL in fasting, after 30 min, 60 min and 90 min, respectively. All the timed growth hormone values were normal except the value at 30 mins which was also not significantly low. Hence GHD was ruled out. Ultrasonography revealed presence of normal uterine structures but her ovaries could not be visualised. MRI of the pelvis confirmed the same with uterus measuring 22×10 mm. MRI of the brain was also normal with pituitary measuring 8×4.4 mm. Echo reports showed normal cardio functions.
Clinical findings of the turner mosaic isochromosome patient.
| S. No. | Particulars | Result |
|---|
| 1 | Karyotype | mos 45,X[6]/47,X,i(X)(q10),i(X)(q10)[6]/46,X,i(X)(q10)[88] |
| 2 | Age (Y) | 6 |
| 3 | Sex | Female |
| 4 | Height (cm) | 85 |
| 5 | Weight (Kg) | 11 |
| 6 | Turner features | Short stature | Yes |
| 7 | Cubitus valgus | No |
| 8 | Webbed neck | No |
| 9 | Wide spaced nipples | No |
| 10 | Shield chest | No |
| 11 | Ultrasonography | Uterus | Normal |
| 12 | Ovaries | Not visualised |
| 13 | MRI-Pelvis | Uterus | Normal; 22×10 mm |
| 14 | Ovaries | Not visualised |
| 15 | Endocrine profile | Total T3 | 2.26 ng/mL |
| 16 | Total T4 | 12.6 μg/dL |
| 17 | TSH | 1.72 μIU/mL |
| 18 | FSH | 8.90 mIU/mL ↑ |
| 19 | LH | 0.04 mIU/mL |
| 20 | Estradiol | <5.0 pg/mL ↓ |
| 21 | Cortisol | 28 μgm/dL ↑ |
| 22 | Growth hormone | Fasting | 0.92 ng/mL |
| 23 | 30 min | 1.3 ng/mL ↓ |
| 24 | 60 min | 28 ng/mL |
| 25 | 90 min | 10.2 ng/mL |
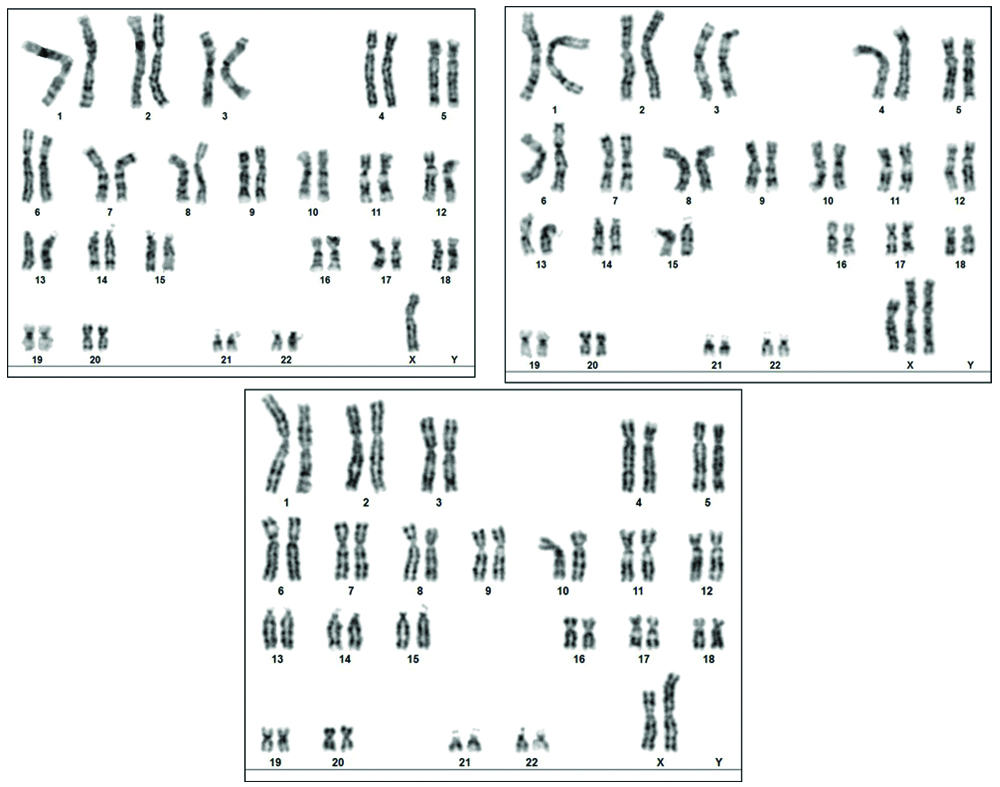
The peripheral blood collected from the proband was subjected to routine chromosomal analysis following the standard protocols. The karyotype was observed to be mos 45,X[6]/47,X,i(X)(q10),i(X)(q10)[6]/46,X,i(X)(q10)[88] and the representative karyogram of the mosaicism is presented in [Table/Fig-3]. The proband’s physical feature, clinical reports and chromosomal data confirms the presence of the TS. The presence of mosaic karyotype further categorises the patient under the Turner variant type. The patient and her parents were given genetic counseling and were recommended hormonal therapy. Since the proband was very young, she was advised for a regular follow-up. This will provide us an insight about prognosis of the Turner features, its associated anomalies and also the fertility status of the patient at the adolescence. The family members didn’t cooperate for cytogenetic testing.
GTG-banded karyogram showing the mos 45,X[6]/47,X,i(X)(q10),i(X)(q10)[6]/46,X,i(X)(q10)[88] karyotype of the proband.
Discussion
Complete or partial deletion of X chromosome results in a condition called Turner syndrome. The missing genetic material affects development and causes the following characteristic features in TS patients. They tend to be shorter than average and are usually infertile because of absence of ovarian function. Turner features include extra skin on the neck (webbed neck), puffiness or swelling (lymphoedema) of the hands and feet, broad chest with widely spaced nipples, skeletal abnormalities, pterigium colli, cubitus valgus, lack of spontaneous pubertal development resulting from ovarian sex hormone insufficiency, low-posterior hairline, misshapen or rotated ears, narrow palate with crowded teeth, hyperconvex nails, multipigmented nevi, cardiac malformation and kidney problems. Diverse pattern of developmental delays, learning disabilities, hearing loss and behavioural problems are observed in these patients [1,2]. Even though the proband in this case report, presented a rare Turner variant karyotype, except short stature none of these significant Turner stigmata’s were observed.
Roughly 3% of the female foetuses are affected by TS and only 1% of them do survive [3]. Hook EB and Warburton D postulated the presence of the critical or second sex chromosome in a 45,X fetus [4], which is very crucial and could be identified in 0-80% of the pure line TS cases when fibroblasts are karyotyped along with molecular screening [3]. Presence of the second cell line in some organs or tissues in the embryonic stage which is later lost due to in vivo selection is hypothesised to be one of the cause and thus the presence of mosaicism either 46,XX or 46,XY becomes indispensable for survival of the foetus [3,4]. The frequency of various karyotypes observed in TS as per literature are 45,X in 50%; 45,X/46,XX in 20%; 46,X,i(Xq) in 15%; 46,X,r(X) or 46,X,del(X) in 10% and remaining 5% of the population expresses other variants [5].
Structural anomalies of X chromosome have a prevalence of 13-20% in patients with TS. Pure line or mosaic for isochromosome is the second most common abnormality in TS patients. Isochromosomes are defined as the presence of structurally abnormal X chromosome caused by the abnormal transverse misdivision of centromere resulting in monosomy for one arm and trisomy for the other. Individuals with i(Xq) show characteristics similar to individuals with classical 45,X such as streak gonads, short stature and about 50% menstruate spontaneously. Autoimmune disorders and deafness are other associated phenotypes [5]. Studies on parental origin of isochromosome suggested that the paternal (64%) contribution is higher when compared to the maternal (36%) [6].
The abnormality was first reported by Margaret M et al., in a 17-year-old patient with provisional diagnosis of primary amenorrhea, who exhibited short stature and streak gonads followed by Gersak K and Gersak ZM in a patient with premature ovarian failure [7,8]. The probands in both the studies were adults but this is the first report in a six-year-old girl child.
The invariable TS feature, the short stature observed in the proband is because of the absence of two copies of genes which is accountable for stature found on the distal short arm (PAR regions) of X (Xp11-22) and Y (Yp11). These genes are found to escape the random inactivation process. SHOX (short stature homeobox-containing gene), or PHOG (pseudoautosomal homeobox containing osteogenic gene) gene located at the PAR1 region, predominantly encodes proteins In bone fibroblasts, but its prime duty is for coding proteins responsible for stature. Expression studies concluded the action of this gene in Y chromosomes and in both the active and inactive X chromosome. Skeleton development, growth and maturation of bones in arms and legs are influenced by the SHOX proteins and thus mutations of it results in short stature. Haplo-insufficiency of this gene is responsible for the bone and skeletal abnormalities since both the copies of the SHOX gene is crucial [9].
Ovarian failure in TS is reported in approximately 95% of the patients, which occurs as a result of the haplo-insufficiency of ovarian genes that escapes inactivation. Until 14 weeks of gestation, TS foetus contain normally appearing ovaries, but the absence of the second active copy or abnormal X chromosome results in decrease in the follicles number due to degeneration or atresia. Accelerated germ cell apoptosis usually occurs at around 18 weeks of gestation in TS patients [2]. The time and rate at which the ovarian failure occurs varies with every TS individual owing to the patient’s cell line. The Xp11, Xq13-25, and Xq26-28 are regarded as “critical regions” for oocyte development, maturation and preservation. ZFX (zinc finger) mapped to distal Xp and DFFRX (Drosophila fat facets related X) to Xp11. Four are other genes responsible for maintenance of ovarian and gonadal function respectively. When these genes are ablated in animal models, it resulted in accelerated ovarian failure and gonadal failure correspondingly [2,10] which is most common manifestation of the Turner mosaic patients. Of the carriers of deletion of short arm or long arm of X about 65% and 93%, respectively, suffer from ovarian dysgenesis. Some cases of monosomy of short arm of X chromosome have normal ovarian function and are fertile [11]. Even though the proband had normal uterine structure, its functional status can be only evaluated at her pubertal age.
Marzuki NS et al., proposed an increased risk for hypothyroidism, autoimmunity, particularly thyroiditis, inflammatory bowel disease, deafness and Intellectual Disability (ID) in patients with isochromosome when compared with the monosomy X population due to duplication of XIST region [12]. Other clinical malformations related to isochromosome were oedema, cardiac and renal anomalies. Deletions of Xp rarely contribute to ID and autism [13]. Subjects missing distal Xp22.33 manifested the TS-associated neurocognitive profile [14]. However, none of these conditions were found in the index patient.
Conclusion(s)
This article emphasises the importance of conventional cytogenetic study in patients with short stature. Early recognition and diagnosis of Turner’s syndrome, alike this case will aid in the management of the disease and the quality of the patient’s life.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes (Taken from Guardian)
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Nov 08, 2019
Manual Googling: Dec 04, 2019
iThenticate Software: Dec 24, 2019 (9%)
[1]. Turner HH, A syndrome of infantilism, congenital webbed neck, and cubitus valgusEndocrinology 1938 23(5):566-74.10.1210/endo-23-5-566 [Google Scholar] [CrossRef]
[2]. Goetsch AL, Kimelman D, Woodruff TK, Disorders of the Sex Chromosomes and Sexual Development. In: Goetsch AL, Kimelman D, Woodruff TK, editorsFertility Preservation and Restoration for Patients with Complex Medical Conditions [Internet] 2017 ChamSpringer International Publishing:19-37.10.1007/978-3-319-52316-3_3 [Google Scholar] [CrossRef]
[3]. Djordjević VA, Jovanović JV, Pavković-Lučić SB, Drakulić DD, Djurović MM, Gotić MD, Cytogenetic findings in Serbian patients with Turner’s syndrome stigmataGenet Mol Res 2010 9(4):2213-21.10.4238/vol9-4gmr95321064029 [Google Scholar] [CrossRef] [PubMed]
[4]. Hook EB, Warburton D, The distribution of chromosomal genotypes associated with Turner’s syndrome: live birth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicismHum Genet 1983 64(1):24-27.10.1007/BF002894736683706 [Google Scholar] [CrossRef] [PubMed]
[5]. Akbas E, Altintas ZM, Celik SK, Dilek UK, Delibas A, Ozen S, Rare types of turner syndrome: clinical presentation and cytogenetics in five casesLab Med 2012 43(5):197-204.10.1309/LMEZQXK85CDP4HYN [Google Scholar] [CrossRef]
[6]. Sagi L, Zuckerman-Levin N, Gawlik A, Ghizzoni L, Buyukgebiz A, Rakover Y, Clinical significance of the parental origin of the X chromosome in turner syndromeThe Journal of Clinical Endocrinology & Metabolism 2007 92(3):846-52.10.1210/jc.2006-015817192299 [Google Scholar] [CrossRef] [PubMed]
[7]. Margaret M, Tilak P, Rajangam S, 45,X/47,X, i(X)(q10),i(X)(q10)/46,X,i(X)(q10) Isochromosome Xq in mosaic turner syndromeInt J Hum Genet 2010 10(1-3):77-80.10.1080/09723757.2010.11886088 [Google Scholar] [CrossRef]
[8]. Gersak K, Gersak ZM, Chromosomal abnormalities and menstrual cycle disorders. chromosomal abnormalities- A hallmark manifestation of genomic instability [Internet] 2017 3010.5772/67795PMC5712847 [Google Scholar] [CrossRef] [PubMed]
[9]. Fukami M, Seki A, Ogata T, SHOX Haploinsufficiency as a cause of syndromic and nonsyndromic short statureMol Syndromol 2016 7(1):03-11.10.1159/00044459627194967 [Google Scholar] [CrossRef] [PubMed]
[10]. Vijayalakshmi SR, Aggarwal S, Genetics of premature ovarian failureGenetic Clinics 2018 11(2):10-17. [Google Scholar]
[11]. Bispo AV, Dos Santos LO, Burégio-Frota P, Galdino MB, Duarte AR, Leal GF, Effect of chromosome constitution variations on the expression of Turner phenotypeGenet Mol Res 2013 12(4):4243-50.10.4238/2013.March.13.1323546984 [Google Scholar] [CrossRef] [PubMed]
[12]. Marzuki NS, Anggaratri HW, Suciati LP, Ambarwati DD, Paramayuda C, Kartapradja H, Diversity of sex chromosome abnormalities in a cohort of 95 Indonesian patients with monosomy XMol Cytogenet 2011 4:2310.1186/1755-8166-4-2321992692 [Google Scholar] [CrossRef] [PubMed]
[13]. Joost K, Tammur P, Teek R, Zilina O, Peters M, Kreile M, Whole Xp deletion in a girl with mental retardation, epilepsy, and biochemical features of OTC deficiencyMol Syndromol 2011 1(6):311-15.10.1159/00033132322190902 [Google Scholar] [CrossRef] [PubMed]
[14]. Ross J, Zinn A, McCauley E, Neurodevelopmental and psychosocial aspects of Turner syndromeMent Retard Dev Disabil Res Rev 2000 6(2):135-41.10.1002/1098-2779(2000)6:2<135::AID-MRDD8>3.0.CO;2-K [Google Scholar] [CrossRef]