Splenic Marginal Zone Lymphoma: A Case with Long History
Michael Leonard Anthony1, Harish Chandra2, Arvind Gupta3, Uttam Kumar Nath4, Ashok Singh5
1 Senior Resident, Department of Pathology, AIIMS, Rishikesh, Uttarakhand, India.
2 Additional Professor, Department of Pathology, AIIMS, Rishikesh, Uttarakhand, India.
3 Associate Professor, Department of Pathology, AIIMS, Rishikesh, Uttarakhand, India.
4 Additional Professor, Department of Hematology and Medical Oncology, AIIMS, Rishikesh, Uttarakhand, India.
5 Assistant Professor, Department of Pathology, AIIMS, Rishikesh, Uttarakhand, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Ashok Singh, Department of Pathology, Second Floor, Medical College Block, AIIMS, Veerbhadra Road, Rishikesh-249201, Dehradun, Uttarakhand, India.
E-mail: ashok.path@aiimsrishikesh.edu.in
Splenic Marginal Zone Lymphoma (SMZL) is a rare, indolent B-cell Non-Hodgkin Lymphoma (NHL) which may present with isolated splenomegaly. Herein, we report a case of SMZL in a 48 years old male with asymptomatic pancytopenia and insidious development of massive splenomegaly over a period of 11 years, where splenectomy not only revealed the diagnosis of SMZL after bone marrow biopsy was non-diagnostic, but also resulted in normalisation of blood counts without any further treatment.
Non-hodgkin lymphoma, Pancytopenia, Splenomegaly
Case Report
A 48-year-old male patient presented to Surgery department with recent onset of early satiety and history of slowly progressive left-sided abdominal fullness of 11 years duration. There was no history of fever, weight loss, night sweats, vomiting, gastrointestinal or muco-cutaneous bleeding, or recurrent infections. He had no co-morbid illness. There was no history of jaundice, tuberculosis, or blood product transfusion, and patient had no addiction to smoking or alcohol.
On systemic examination he had mild pallor and massive splenomegaly (spleen palpable 10 cm below left costal margin), but no jaundice, sternal tenderness, peripheral lymphadenopathy, hepatomegaly, ascites, tonsillar or testicular enlargement, any skin lesion, or cranial nerve palsy. Complete haemogram revealed pancytopenia (haemoglobin 9.6 g/dl, platelet count 70×109/L, and total leukocyte count 3.1×109/L, with 37% neutrophils, 56% lymphocytes, 5% monocytes and 2% eosinophils). Reticulocyte count was 2.5%. Peripheral smear showed microcytic hypochromic red blood cells and no abnormal lymphoid cells were seen. Direct Coombs test was done to rule out auto-immune haemolytic anemias and it was negative. Coagulation profile (APTT, PT, thrombin time, fibrinogen level), haemoglobin HPLC, renal and liver function tests, electrolytes and serum LDH level were normal. HBsAg was negative and anti-HIV and anti-HCV were non-reactive. Abdominal ultrasound showed massive splenomegaly without any feature of portal hypertension. Gastro-duodenoscopy findings were normal.
A provisional diagnosis of hyperspleenism was made with a differential diagnosis of malaria and leishmania. Bone marrow aspiration and biopsy which were done in another laboratory prior to presentation were reported as normal.
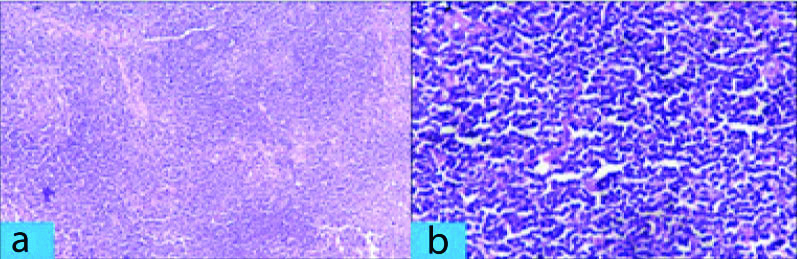
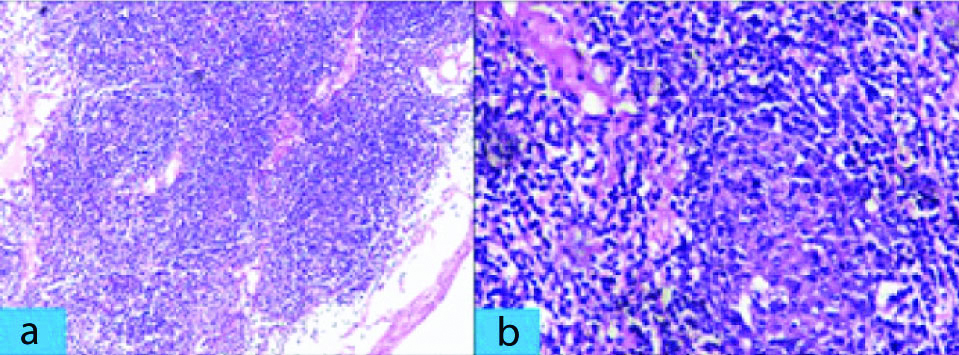
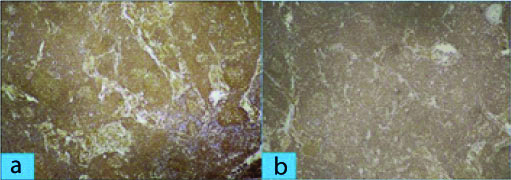
The patient underwent splenectomy in Surgery department for persistent abdominal discomfort. The splenectomy specimen measured 24×19×11 cm and weighed 3.1 kg, and included multiple splenic hilar lymph nodes (size ranging between 0.5×0.5×0.5 cm and 2×2×1 cm). Sections from spleen showed thickened fibrous capsule along with enlarged white pulp of varying sizes. The follicles of the white pulp [Table/Fig-1a,b] showed expansion of marginal zone by small lymphocytes and formation of residual germinal centers along with infiltration by atypical cells in red pulp. Sections from splenic hilar lymph nodes [Table/Fig-2a,b] showed effacement of nodal architecture by atypical small lymphocytes which had scant cytoplasm, and slightly irregular nuclei with inconspicuous nucleoli. After histopathological examination, a diagnosis of NHL with a differential of SMZL lymphoma, small lymphocytic lymphoma, mantle cell lymphoma, follicular lymphoma was made. Immunohistochemistry (IHC) was done to confirm and further subtype Hodgkin lymphoma since on H&E, a diagnosis of non-hodgkin was made. The tumor cells were positive for CD20 and negative for CD5, CD10, CD23, CD43, BCL2, BCL6 and cyclin D1 [Table/Fig-3a,b]. The overall features were consistent with diagnosis of SMZL.
Section from spleen. a) Lymphoid cells which are small, monocytoid and monotonous. (H&E Stain X 400); b) Obliteration of splenic parenchyma due overgrowth of lymphoid cells in marginal zone of the white pulp. (H&E Stain X 100).
Section from hilar lymph nodes. a) Expansion of marginal zone with obliteration of normal lymph node architecture (H&E Stain X 100); b) Atypical small lymphocytes which have scant cytoplasm and slightly irregular nuclei (H&E Stain X 400).
a) Section from spleen showing atypical lymphoid population of cells strongly staining for CD 20. (IHC X 100); b) Section from hilar lymph nodes showing atypical lymphoid population of cells strongly staining for CD 20. (IHC X 100).
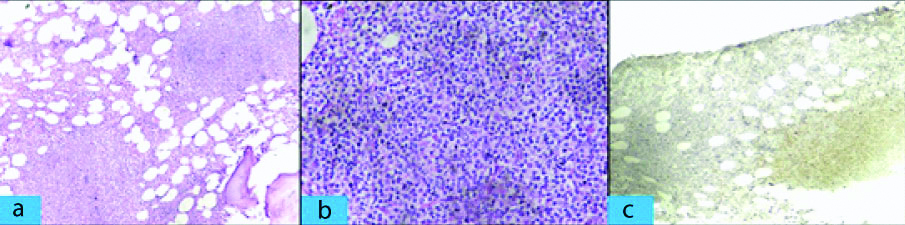
A repeat bone marrow biopsy showed mild erythroid hyperplasia and few lymphoid aggregates which were CD20 positive [Table/Fig-4a-c]. Bone marrow cytogenetic study revealed normal karyotype. Serum protein electrophoresis and immunofixation were negative, and quantitative immunoglobulin levels were normal. There was complete resolution of pancytopenia after three weeks of splenectomy. The patient is asymptomatic and blood counts were normal after two years of follow-up.
Section from bone marrow. a) Infiltration by atypical lymphoid cells forming nodular aggregates in bone marrow. (H&E Stain X 100); b) Atypical lymphoid cells with irregular nuclei. (H&E Stain X 400); c) Atypical lymphoid population of cells to be strongly staining for CD 20 (IHC X 100).
Discussion
SMZL is a rare, indolent small B-cell lymphoma comprising <1% of NHLs [1]. Patients present with moderate to massive splenomegaly. Bone marrow is frequently involved but peripheral lymphadenopathy is uncommon [2]. Spleen histology is the gold standard to establish the diagnosis of SMZL.
Around 8-14% of surgical splenectomies done for lympho-proliferative disorder make-up for this lymphoma [3]. Male to female ratio is 1:1.8 and a median age of these patients is around 68 years (range, 22-79 years) [4]. SMZL involves the spleen in all cases and splenic hilar lymph nodes are often involved, but peripheral lymph node are usually not involved. Bone marrow involvement is seen in 85% of patients, and peripheral blood involvement is seen in 30-50% of patients. SMZL is most definitively diagnosed by splenectomy, because bone marrow morphology may not be diagnostic and immunophenotype is non-specific. Typical immunophenotype is CD20(+), CD5(-), CD10(-), CD23(-/+), CD43(-/+) and cyclin D1(-) [1]. The clinical course is usually indolent, with 10-year survival probability of 67-95% [5]. Asymptomatic patients are observed until they develop indications of treatment. The indications of treatment in SMZL are progressive cytopenia and disease-related symptoms (symptoms due to splenomegaly, weight loss, early satiety, abdominal pain). Treatment options for symptomatic patients are splenectomy or Rituximab [1]. Splenectomy alone can result in overall response rates of 80-90%, with median overall survival of 93 months as reported in retrospective series [6]. At present, the preferred first line treatment for symptomatic patients is Rituximab monotherapy (375 mg/m2 weekly × 4 doses), and splenectomy is indicated if there is no response to Rituximab [1,7,8]. In the present case, splenectomy not only provided the diagnosis of SMZL after the initial bone marrow biopsy was non-diagnostic, but also resulted in complete recovery of pancytopenia.
Immunohistochemistry played a major role as cyclin D1 and CD5 were negative which was helpful to rule out Mantle cell lymphoma, CD10 and BCL6 were negative which helped to exclude Follicular lymphoma and absence of CD5 and CD23 to rule out small lymphocytic lymphoma. There is not much of a change in SMZL according to WHO 2017 classification. The presence of NOTCH2, KLF2 and TP53 mutations are associated with an unfavourable outcome [9].
This case report also highlights that a lymphoma should always be considered in the differential diagnosis of a pancytopenia patient with massive splenomegaly not responding to treatment in tropical countries where infectious diseases are generally thought off.
Conclusion
The present case report acts as a reminder that splenectomy can provide definitive diagnosis of SMZL in patients with unexplained splenomegaly and pancytopenia, even if bone marrow study is non-diagnostic. It also highlights the fact that indolent lymphomas like SMZL can progress insidiously over unusually long periods of time and a high index of clinical suspicion is warranted for accurate diagnosis, in order to avoid unnecessary treatment.
Author Declaration:
Financial or Other Competing Interests: No
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. NA
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Sep 10, 2019
Manual Googling: Nov 31, 2019
iThenticate Software: Nov 20, 2019 (14%)
[1]. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. B-Cell Lymphomas, Version 5.2019 – September 23, 2019. www.NCCN.org [Google Scholar]
[2]. Jamal I, Nirala S, Tutor P, Splenic marginal zone lymphoma: A rare case report on splenic marginal zone lymphomaIOSR-JDMS 2016 15(9):38-40.10.9790/0853-1509113840 [Google Scholar] [CrossRef]
[3]. Mishra MN, Pandey R, Dinda A, Nityanand S, A peculiar case of splenic marginal zone lymphoma and review of literatureIran J Immunol 2013 10(3):186-89. [Google Scholar]
[4]. Franco V, Florena AM, Iannitto E, Splenic marginal zone lymphomaBlood 2003 101(7):2464-72.10.1182/blood-2002-07-221612446449 [Google Scholar] [CrossRef] [PubMed]
[5]. Lenglet J, Traullé C, Mounier N, Benet C, Bongrand NM, Amorin S, Long-term follow-up analysis of 100 patients with splenic marginal zone lymphoma treated with splenectomy as first-line treatmentLeuk Lymphoma 2014 55(8):1854-60.10.3109/10428194.2013.86106724206091 [Google Scholar] [CrossRef] [PubMed]
[6]. Milosevic R, Todorovich M, Balint B, Jevtic M, Krstic M, Ristanovic E, Splenectomy with chaemotherapy vs surgery alone as initial treatment of splenic marginal zone lymphomaWorld J Gastroenterol 2009 15(32):4009-15.10.3748/wjg.15.400919705496 [Google Scholar] [CrossRef] [PubMed]
[7]. Tsimberidou AM, Catovsky D, Schlette E, O’Brien S, Wierda WG, Kantarjian H, Outcomes in patients with splenic marginal zone lymphoma and marginal zone lymphoma treated with rituximab with or without chemotherapy or chemotherapy aloneCancer 2006 107(1):125-35.10.1002/cncr.2193116700034 [Google Scholar] [CrossRef] [PubMed]
[8]. Else M, Marin-Niebla A, de la Cruz F, Batty P, Rios E, Dearden CE, Rituximab, used alone or in combination, is superior to other treatment modalities in splenic marginal zone lymphomaBr J Haematol 2012 159(3):322-28.10.1111/bjh.1203623016878 [Google Scholar] [CrossRef] [PubMed]
[9]. Piris MA, Isaacson PG, Swerdlow SH, Thieblemont C, Pittaluga S, Rossi D, Spleenic B cell marginal zone lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. (Eds)WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues 2017 Revised 4th edFranceInternational Agency for Research on Cancer (IARC), Lyon:225 [Google Scholar]