Leigh’s syndrome or sub-acute necrotizing encephalopathy is a rare progressive neurodegenerative disorder of paediatric age group. It has variable clinical, imaging and pathological presentation. Typical MRI features lead to the diagnosis. It is usually of infantile or juvenile form, which presents in <5 years of age. Here, the present authors report an unusual case of 7-year-old male presented with complaints of frequent falls, slurring of speech, difficulty in walking and regression of the achieved milestone was noted. MRI brain study showed altered signal intensity in bilateral putamen, bilateral caudate nucleus, midbrain, substantia nigra, periaqueductal region and pons. Magnetic Resonance spectroscopy revealed high lactate levels in pontine lesions. MRI findings were suggestive of subacute necrotizing encephalopathy.
Case Report
A 7-year-old male born of second-order to non-consanguineous parents, with uneventful perinatal history and normal development for age presented with complaints of frequent falls, difficulty in walking and getting up from sitting posture since a period of four months. He also presented with progressive cognitive dysfunction and slurring of speech over the past four months during which regression of the achieved milestone was noted.
On examination, there was microcephaly along with mild scoliosis. Neurological examination revealed increased tone with hyperactive deep tendon reflexes, bilateral positive extensor plantar response and clonus. Routine investigation comprising of complete blood count, urinalysis, liver and renal functions along with triglyceride, cholesterol, and uric acid levels were performed and found to be under normal limits. Arterial blood gas analysis revealed mild metabolic acidosis, with normal anion gap. Arterial lactate levels and total Creatine Phosphokinase (CPK) levels were performed which revealed raised values. Serum lactate was 3.18 mmol/L (reference range- 0.5-2.2). However, blood ammonia levels and CPK-MB levels were within normal limits.
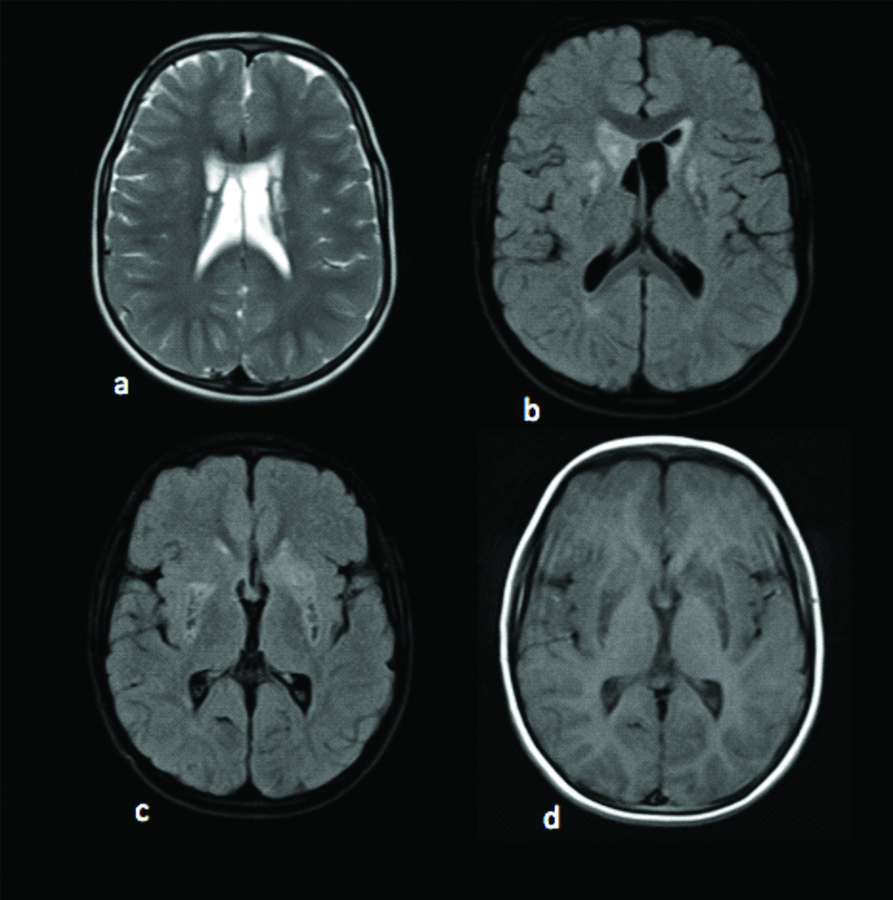
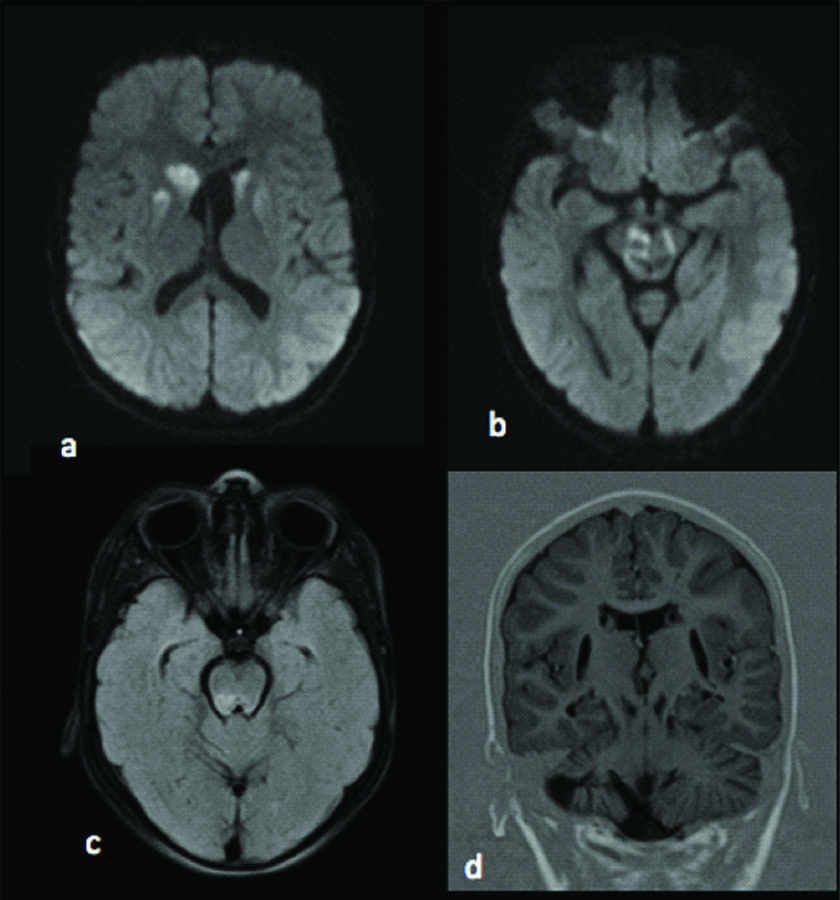
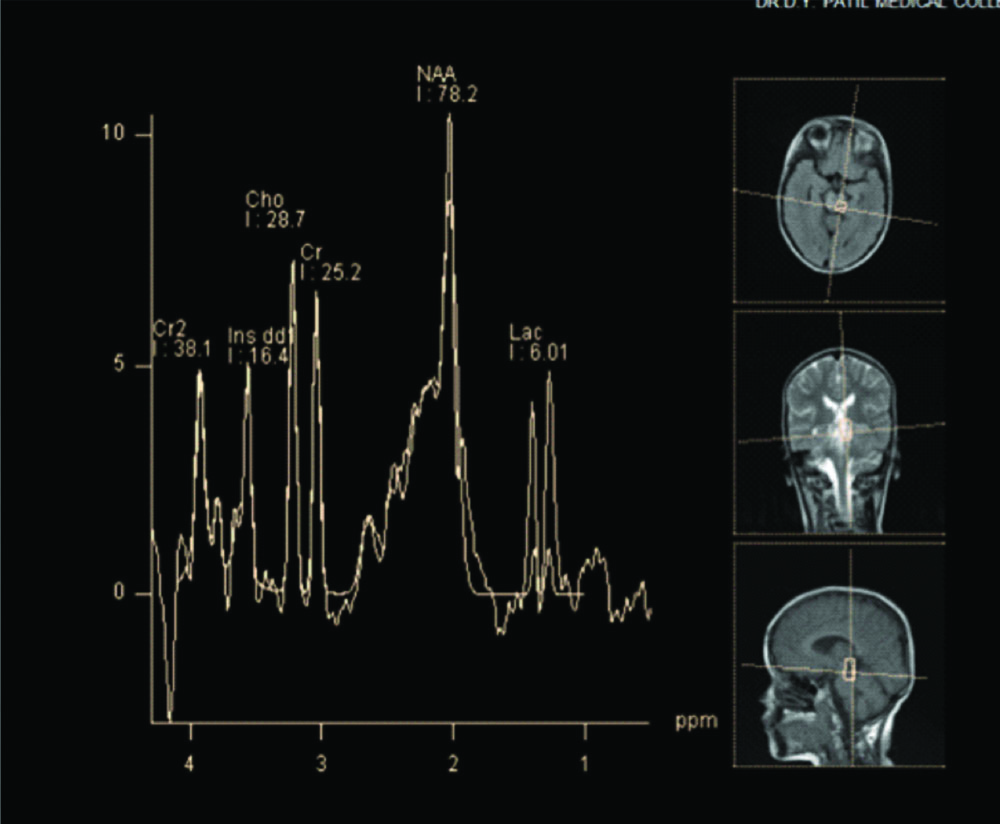
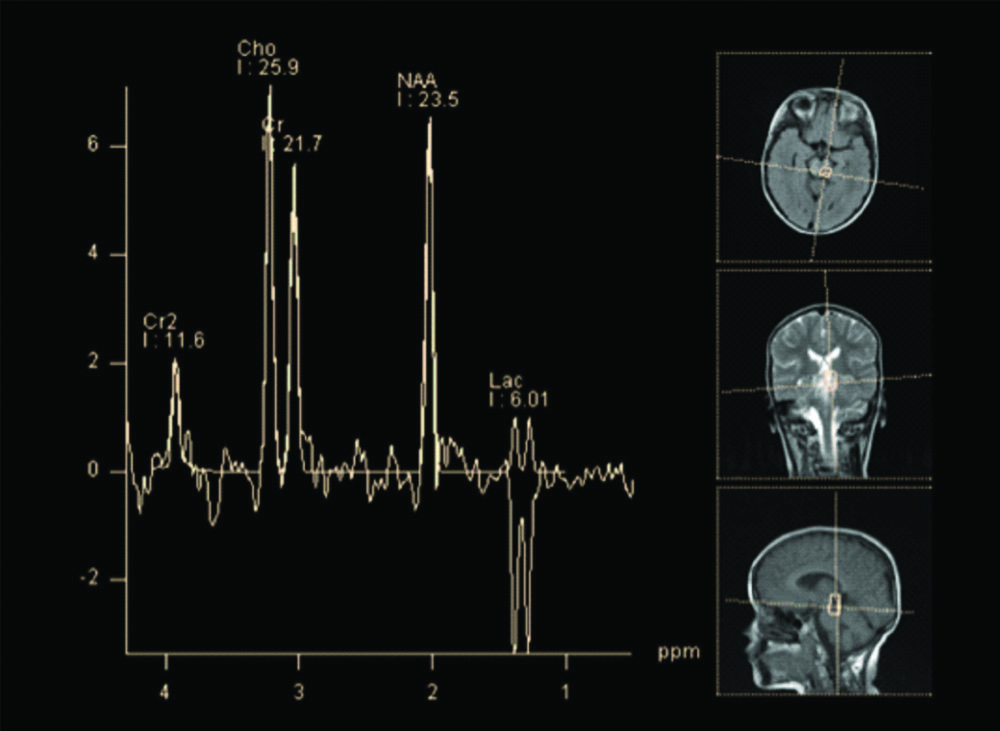
Electroencephalogram (EEG) showed slowing of background rhythm with no epileptic activity. Fundus examination was unremarkable. MRI brain was obtained with spectroscopy. MRI revealed altered signal intensities in bilateral putamen, bilateral caudate nucleus predominantly on right side, midbrain, substantia nigra, periaqueductal region and pons which appeared hyperintense on T2WI, hypointense on T1WI and heterogeneously hyperintense on FLAIR with restriction on DWI [Table/Fig-1,2]. Mild cortical atrophy of right cerebellar hemisphere was also noted. MR spectroscopy revealed high choline level in basal ganglia and pontine lesions and high lactate levels in pontine lesions. An elevated doublet lactate peak was found at an echo time of 135 ms (downward peak at this TE Time), reduced NAA peak suggested loss of neuronal integrity [Table/Fig-3,4].
MRI axial images: a) T2WI showing altered signal intensity in the bilateral basal ganglia; b,c) Axial FLAIR images demonstrating hyperintense signal intensity with hypointense necrotic areas in bilateral putamen, bilateral caudate nucleus; d) Axial T1W image showing hypointense signal intensities in bilateral gangliocapsular region.
a,b) Axial DWI images demonstrating diffusion restriction in the bilateral thalami, basal ganglia, mid brain and substantia nigra; c) Axial FLAIR image showing hyperintense signals in pons; d) Coronal image in Inversion recovery showing atrophy of right cerebellum.
MR Spectroscopy done from the affecting region of pons revealed an increase in the Lactate doublet peak at echo time of 135 ms (Lactate peak is upwards at this TE time).
MR Spectroscopy done from the affecting region of caudal pons revealed a increase in the Choline peak. An elevated doublet lactate peak is found at echo time of 135 ms (downward peak at this TE Time). Reduced NAA peak suggested loss of neuronal integrity at the same region.
Molecular studies and enzymology could not perform due to financial constraints. The child was treated conservatively to improve the ATP production with ketogenic diet and thiamine supplement. Physiotherapy was given for the neuromuscular symptoms.
Discussion
Leigh’s syndrome, a mitochondrial disorder, is a rare progressive neurodegenerative disorder presenting in the infancy or childhood. It is also termed as sub-acute necrotizing encephalopathy. Only few cases were reported from India. Denis Leigh, British neuro-pathologist reported the first case in 1951 [1]. Although rare; Leigh syndrome is the most common mitochondrial disease presenting in childhood with an incidence of 1:40,000 live birth [2]. Leigh syndrome has varied presentation including psychomotor delay or regression, ataxia, hypotonia, dystonia, gait disturbance, seizures, respiratory insufficiency, pyramidal signs and lactic academia. These symptoms worsen gradually [2,3]. Respiratory chain enzyme dysfunction following defect in the genes contribute to majority of the cases [4]. Dysfunction in the respiratory chain enzymes is responsible for the disease in most of the cases. Pattern of inheritance may be X-linked recessive, autosomal recessive or mitochondrial. Genetic or biochemical abnormality cannot be found in 35% to 60% cases [2]. Therefore, sub-acute necrotizing encephalopathy is better diagnosed on the basis of their clinical signs, symptoms, mode of transmission, metabolic abnormalities and neuroimaging [4].
Neuroimaging with MRI along with MR spectroscopy of the affected area has proved to be of great importance in the early diagnosis. MR Spectroscopy is a non-invasive method to measure and monitor the biochemical changes in various lesions. Each metabolite in the brain affects specific cellular and biochemical processes and appears at a specific ppm. Hence the observed MR spectroscopy finding of a lesion as along with MRI finding correlation gives more information about the disorder being evaluated.
Sub-acute Necrotizing Encephalomyelopathy (SNE) presents broadly in two forms, infantile and juvenile form. Within two years of life, most of the cases of infantile form of Leigh’s syndrome has characteristic clinical features of breathing dysfunctions, ataxia, seizures, regression of neuro-psychomotor development and hypotonia [5,6]. In the juvenile form (less common), most of the patients present with extrapyramidal syndrome with dystonia and stiffness. This syndrome is genetically related to an enzyme defect in any part of the cell respiration, and the most common being biochemical deficiencies of pyruvate dehydrogenase complex, complex IV (or cytochrome C oxidase), in the Complex I of respiratory chain or in the ATPase [4-7]. Several studies showed significant heterogeneity in the clinical findings of Leigh syndrome. Sofou K et al., revealed in a study that 69% of the cases presented before 2 years of age, while three patients presented with ataxia or seizures at 8,10 and 21 years of age [8]. In the index case, the child presented at the age of 7 years with ataxia, gait disturbances and neuroregression. Adult onset Leigh disease is designated to patients who survive longer than 18 years. Adult onset Leigh disease has lesser incidence of serum lactate elevation, basal ganglia lesion, COX deficiency and developmental delay. The patients present with cranial nerve disorders, pyramidal signs and cerebellar dysfunction [9].
Leigh’s syndrome may be autosomal recessive or X-linked or mitochondrial in inheritance. Diagnosis of this syndrome is suspected by MRI in most of the cases, especially in late or atypical forms. Basal ganglia and brain stem are the most commonly affected areas. MRI findings include symmetric hyperintense lesions of basal ganglia especially putamen and globus pallidus, brain stem and thalamus on T2W images [4,10,11]. Cerebral cortex and cerebellum are less commonly involved. Very rare involvement of white matter of the brain has also been reported. The case presented here describes an atypical form in terms of the age of presentation, which falls under the juvenile form of Leigh disease and showed a gradual progression in symptoms which seem to occur in the late form of presentation. Diagnosis was made by the association of clinical manifestations with the findings on MRI brain and MR Spectroscopy.
MRI imaging revealing a pattern of symmetrical, hyperintense lesion on T2 weighted image involving the basal ganglia and brainstem, with predominant involvement of putamen, is highly specific for SNE [4].
Pathologically, the changes in the Leigh syndrome is characterised by multifocal spongiform lesions with relative preservation of neurons associated with degeneration and gliosis through the brain, including basal ganglia, brain stem, thalami, cerebellum, spinal cord and optic nerves [2].
MR imaging helps in differentiating the other pathological conditions affecting the basal ganglia manifesting in the childhood which are not well differentiated on CT brain [4].
MR spectroscopy provides information about the pathophysiology of the disease and helps to assess the diagnosis. Studies have reported an increase in lactate at MR spectroscopy in affected areas of the patient, in the basal ganglia with mitochondrial disease [7,12,13].
Barkovich AJ et al., reported an increase in lactate from the affected areas of patients, on MR spectroscopy [14]. MR Spectroscopy findings, clinical symptoms and laboratory findings are altogether important for diagnosing Mitochondrial Encephalopathies.
There is wide heterogeneity of the clinical features of the Leigh syndrome and notable absence of the specific biochemical or molecular defect, diagnosis usually relied on typical MRI brain features and clinical findings [2,15].
Diffusion weighted MRI show restriction in acute stages. MR Spectroscopy show decreased level of N-acetyl aspartate peak and elevated lactate peak. Decrease in NAA level occurs due to the neuronal and/or axonal loss and necrosis. Lactate peak is feature of dysfunctional OXPHOS [16]. In the present case, the affected areas had decreased NAA and lactate peak. However, demonstration of symmetric basal ganglia, brain stem and subthalamic nuclei involvement in MRI indicates a mitochondrial disorder in children, the combination of MR spectroscopy and brain MRI increases the specificity for the detection of LS especially for the juvenile form [14,17,18].
Most of the previous case reports are of younger children (<5 years) but in this case, the age of the patient was 7 years, which was unusual age of presentation. Other mitochondrial diseases, hypoxic encephalopathy, Wilson’s disease and lactic acidosis can show basal ganglia lesions and considered to be differentials.
Specific treatment for the Leigh syndrome is not available and available therapeutic options are limited. Symptomatic treatment is usually given to improve the ATP production and to lower the lactate levels. Ketogenic diet is found to improve the clinical features [16]. Supplements with thiamine and physiotherapy are recommended for the neuromuscular concerns. Antiseizure medication is suggested for seizure control. Response to the treatment can be monitored by MRI Brain and MRS.
Conclusion
Magnetic resonance imaging with modern imaging techniques and MR spectroscopy is a valuable investigation for the extent of the disease and diagnosis of subacute necrotizing encephalomyelopathy and for the evaluation of its outcome.
[1]. Leigh D, Subacute necrotizing encephalomyelopathy in an infantJ Neurol Neurosurg Psychiatry 1951 14:216-21.10.1136/jnnp.14.3.21614874135 [Google Scholar] [CrossRef] [PubMed]
[2]. Rahman S, Blok RB, Dahl HH, Leigh syndrome: clinical features and biochemical and DNA abnormalitiesAnn Neurol 1996 39(3):343-51.10.1002/ana.4103903118602753 [Google Scholar] [CrossRef] [PubMed]
[3]. Finsterer J, Leigh and Leigh-like syndrome in children and adultsPediatr Neurol 2008 39:223-35.10.1016/j.pediatrneurol.2008.07.01318805359 [Google Scholar] [CrossRef] [PubMed]
[4]. Medina L, Chi L, De Vivi DC, MR Findings in patients with subacute Necrotizing Encephalomyelopathy (Leigh syndrome)AJNR 1990 154:1269-74.10.2214/ajr.154.6.21596892159689 [Google Scholar] [CrossRef] [PubMed]
[5]. Montpetit VJA, Andermann F, Carpenter S, Fawcett JS, ZborowskaSluis D, Giberson HR, Subacute narcotizing encephalomyelopathyBrain 1971 94:01-30.10.1093/brain/94.1.15552162 [Google Scholar] [CrossRef] [PubMed]
[6]. Sparaco M, Bonilla E, DiMauro S, Powers J, Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defectsJ Neuropathol Exp Neurol 1993 52:01-10.10.1097/00005072-199301000-000018426185 [Google Scholar] [CrossRef] [PubMed]
[7]. Lee H-F, Tsai C-R, Chi C-S, Lee H-J, Chen CC-C, Leigh syndrome: clinical and neuroimaging follow-upPediatr Neurol 2009 40:88-93.10.1016/j.pediatrneurol.2008.09.02019135620 [Google Scholar] [CrossRef] [PubMed]
[8]. Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, A multicenter study on Leigh syndrome: disease course and pre- dictors of survivalOrphanet J Rare Dis 2014 9:5210.1186/1750-1172-9-5224731534 [Google Scholar] [CrossRef] [PubMed]
[9]. Jabeen SA, Sandeep G, Mridula KR, Meena AK, Borgohain R, Sundaram C, Adult-onset Leigh’s disease: A rare entityAnnals of Indian Academy of Neurology 2016 19(1):14010.4103/0972-2327.17543727011650 [Google Scholar] [CrossRef] [PubMed]
[10]. Arii J, Tanabe Y, Leigh Syndrome: Serial MR Imaging and Clinical Follow-upAJNR Am J Neuroradiol 2000 21:1502-09. [Google Scholar]
[11]. Davis PC, Hoffman JC, Braun IF, Ahmann P, Krawiecki N, MR of Leigh’s Disease (Subacute Necrotizing Encephalomyelopathy)AJNR 1987 8:71-75. [Google Scholar]
[12]. Sijens PE, Smit GPA, Rödiger LA, Van Spronsen FJ, Oudkerk M, Rodenburg RJ, MR spectroscopy of the brain in Leigh syndromeBrain & Development 2008 30:579-83.10.1016/j.braindev.2008.01.01118329833 [Google Scholar] [CrossRef] [PubMed]
[13]. Aydina H, Kizilgöz V, Hekimoğlu B, Leigh Syndrome: Cranial MRI and MR Spectroscopy FindingsFırat Tıp Dergisi 2011 16(2):102-05. [Google Scholar]
[14]. Barkovich A, Good W, Koch T, Berg B, Mitochondrial disorders: Analysis of their clinical and imaging characteristicsAJNR 1993 14:1119-37. [Google Scholar]
[15]. Jiang YW, Qin J, Yuan Y, Qi Y, Wu XR, Neuropathologic and clinical features in eight Chinese patients with Leigh diseaseJ Child Neurol 2002 17(6):450-52.10.1177/08830738020170061112174968 [Google Scholar] [CrossRef] [PubMed]
[16]. Ruhoy IS, Saneto RP, The genetics of Leigh syndrome and its implications for clinical practice and risk managementThe Application of Clinical Genetics 2014 7:221-34.10.2147/TACG [Google Scholar] [CrossRef]
[17]. Huntsman RJ, Sinclair DB, Bhargava R, Chan A, Atypical presentations of Leigh syndrome: a case series and reviewPediatr Neurol 2005 32:334-40.10.1016/j.pediatrneurol.2004.12.00915866434 [Google Scholar] [CrossRef] [PubMed]
[18]. Ducreux D, Nasser G, Lacroix C, MR diffusion tensor imaging, fiber tracking and single-voxel spectroscopy findings in an unusual MELAS caseAJNR 2005 26:1840-44. [Google Scholar]