A Case of Limb Girdle Muscular Dystrophy Type 2A from India: Copy Number Variation Analysis using Targeted Amplicon Sequencing
Arpan D Bhatt1, Krati Shah2, Apurva Puvar3, Chaitanya G Joshi4, Madhvi Joshi5
1 Ph.D Student, Department of Biotechnology, Hemchandracharya North Gujarat University, Patan, Gujarat, India.
2 Genetic Counselor, Department of Genetics, One-Centre for Rheumatology and Genetics, Vadodara, Gujarat, India.
3 Ph.D Student, Department of Biotechnology, Hemchandracharya North Gujarat University, Patan, Gujarat, India.
4 Director, Department of Biotechnology, Gujarat Biotechnology Research Center, Gandhinagar, Gujarat, India.
5 Joint Director, Department of Biotechnology, Gujarat Biotechnology Research Center, Gandhinagar, Gujarat, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Madhvi Joshi, Department of Science and Technology, Government of Gujarat, MS Building, Block B and D, 6th Floor, GH Road, Sector-11, Gandhinagar-382011, Gujarat, India.
E-mail: jd1-gbrc@gujarat.gov.in
Limb Girdle Muscular Dystrophy-Type 2A (LGMD-2A) is an autosomal recessive disease caused due to mutation in the Calpain-3 (CAPN3) gene, leading to partial or total loss of protein. In India, LGMD-2A is the most prevalence form of the disease accounting for 47% of cases amongst the heterogeneous group. Here, we report a case of 26-year-old female, having difficulty in walking due to proximal muscle weakness since the age of 13 years and had elevated Creatine Phosphokinase (CPK) with abnormal muscle biopsy findings. She was considered for the targeted gene panel based diagnosis with the query of muscular dystrophy. A homozygous exon 17 to 24 deletion was detected in the CAPN3 gene located in the long arm of chromosome 15, which was consistent with the patient’s clinical reports of calpinopathy.
Calpinopathy, Creatine phosphokinase, Muscular disease
Case Report
A 26-year-old female was suspected to have a muscular disease. She was a first child born to non-consanguineous marriage, delivered normally and without any complication. Her two younger sisters had no similar signs. A three-generation pedigree analysis showed no family history of disease. The patient had no history of alcohol consumption, smoking, allergies or medication apart from vitamin supplementation. She started showing difficulty in walking and climbing stairs at the age of 13. At the current stage, she could walk few steps with support. Neurological examination showed normal cranial functions with speech. All superficial and deep tendon reflexes were normal. Motor nerve examination of both lower limbs showed a score of 3/5 in hips, 4/5 in knees and 3/5 in ankles. Dorsiflexion and plantar flexion were weak. However, scoring of 5/5 in shoulder, elbow, and wrist of both upper limbs were recorded [1]. Her serum CPK level was elevated to 3591 U/L compared to normal level of 22-198 U/L. Patient’s muscle biopsy examination from left quadriceps was done at the age of 14 years, which revealed the presence of lobulated fibers and incomplete fascicles. There was no sign of inflammation or vasculitis. Calf hypertrophy was not present. She exhibits slight curvature of the spine. On the basis of muscle involvement in disease, elevated CPK and muscle biopsy findings, patient was suspected to have muscular dystrophy. She was recruited for the molecular diagnosis of muscular dystrophy at the institute. Her clinical examination and genetic counseling were done by a clinician and certified genetic counselor respectively. The details of her previous reports and other relevant information were collected by personal interview and recorded in the physical form. Blood sample in sterile K3-EDTA tube was collected after obtaining patients’ consent.
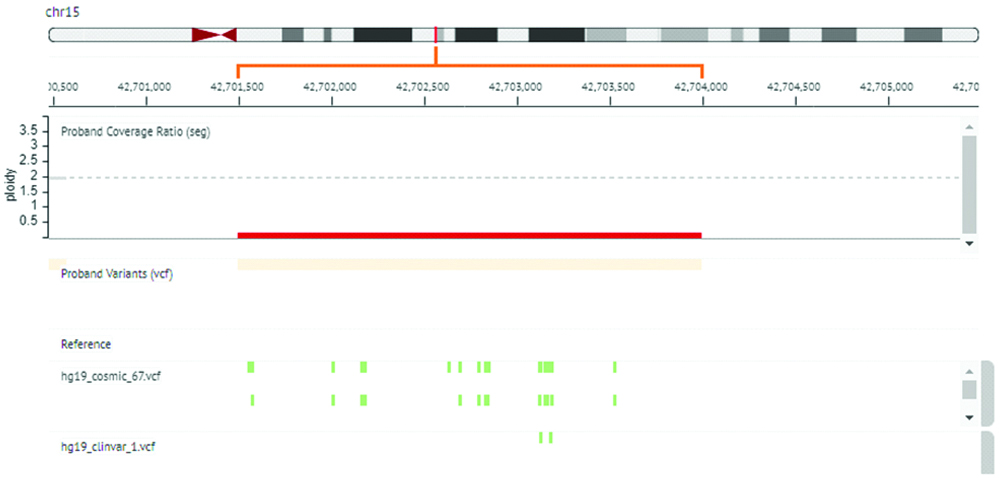
Targeted amplicon sequencing was done for this patient using gene panel for muscular dystrophies on Ion Torrent platform (Ion Personal Genome Machine (PGM) by Thermo Fisher Scientific). Bioinformatics analysis using Ion Reporter version 5.6 for identification of Single Nucleotide Polymorphisms (SNP) was done, however no mutation was detected. Copy Number Variation (CNV) was analysed from data generated by targeted amplicon sequencing of patient’s DNA. For copy number detection, data generated from healthy individuals DNA (n=10) sequencing was used as a reference to create baseline data (hg 19). This baseline data was further used as a reference data line to analyse deletion/duplication in patient sample. Analysis revealed homozygous deletion of exon 17 to 24 of CAPN3 gene in patient [Table/Fig-1].
Copy Number Variation (CNV) analysis result showing homozygous deletion in CAPN3 gene.
*Ion Reporter V.5.6 used for CNV detection in CAPN3 gene. Red bar highlights deleted region of CAPN3 gene with copy number 0. hg19 reference sequence with green spots indicates regions with COSMIC and ClinVar variants reported in deleted region.
Discussion
LGMD-2A is a most common form of Limb Girdle Muscular Dystrophy worldwide, reported with 10 to 33% prevalence rate [2,3]. LGMD-2A inherited in an autosomal recessive manner, is caused by a defect in the CAPN3 gene [3]. LGMD-2A is characterised by a significant deterioration of the muscles of the proximal pelvic and shoulder girdle [4]. Age of onset of this type of LGMD ranges from 2 to 40 years and demonstrates heterogeneity as mild to severe forms [5]. CAPN3 gene found in sarcomere structures of muscle cells, codes for protein “Calpin-3” protein that plays an important role in myofibril integrity and function. The gene maintains “sarcomere” structure and also regulates sarcomeric protein turnover [6]. In India, several hospital-based studies using Immunohistochemistry (IHC) and Western Blotting (WB) as diagnostic tools have reported sarcoglycanopathies, dysferlinopathies and calpinopathies as common LGMDs [7]. Another immunoblotting based Indian study, reported Calpin protein abnormalities in 43.8% LGMD samples analysed, suggesting that calpinopathy is the most common form of LGMD [8]. While the use of the semi-quantitative and histological examination in diagnosis LGMDs is well documented, there is lack of literature on Next Generation Sequencing (NGS) based genetic workups.
Till date, Leiden Muscular Dystrophy Pages (LMDp) database contains 517 unique disease-causing variants of CAPN3 gene. Amongst these c.550 delA mutation is most prevalent reported in Caucasian populations [7,9]. In a recent study by Wang L et al., 38 genetic variations were identified in 24 probands of Asian origin that caused LGMDs, and 18 out of these 38 variations were found to be novel [10]. It shows LGMDs exhibit a high rate of clinical and genetic variation. Often, a detailed clinical workup is required to reach an accurate diagnosis, which is time-consuming and requires the extended presence of patients throughout the procedures. Hence, to avoid multiple procedures of diagnosis, a single screening test was conducted, to identify disease-causing mutation/s in LGMD associated genes. Since the inception of NGS technology per base sequencing cost has reduced drastically and it has emerged as an accurate, in-depth and affordable option for heterogeneous disorders. Several options like Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES) are available to diagnose LGMDs using NGS which provides a complete scenario of human genome and exome respectively. However, to reduce the cost of testing, adopting a targeted approach of sequencing genes responsible for muscular dystrophies using NGS has been adopted.
NGS identified homozygous deletion of exon 17 to 24 in CAPN3 gene, has not been reported previously and hence can be categorized as a novel variant. Exon 17 to 24 is necessary for the translation of amino acids position 639 to 821 of CAPN3. Deletion of these exons leads to short truncated non-functional CAPN3 protein.
The patient’s clinical reports (age of onset, limbs affected, rate of progression), motor nerve examination, biochemical and hisopathological findings suggested end stage muscular dystrophy. Molecular investigation by muscular dystrophy gene panel using NGS identified deletion in CAPN3 gene, which further helped in conclusive diagnosis of calpinopathy.
Conclusion
This study reports a deletion in the CAPN3 gene using target NGS panel in 26-year-old female suspected with muscle disease. Acquiring this non-invasive, cost-effective and rapid approach for the diagnosis of muscular dystrophies should be of priority over other invasive techniques. If results are inconclusive using a targeted approach, screening of mutations in whole exome or immunostaining methods can be reviewed.
Funding: This work was supported by Department of Science and Technology, Government of Gujarat.
[1]. Ouellete H, Orthopedics Made Ridiculously Simple (Medmaster Ridiculously Simple) 2008 Med Master Inc:59 [Google Scholar]
[2]. Murphy AP, Straub V, The classification, natural history and treatment of the limb girdle muscular dystrophiesJ Neuromuscul Dis 2015 2(2):S7-S19.10.3233/JND-15010527858764 [Google Scholar] [CrossRef] [PubMed]
[3]. Taghizadeh E, Rezaee M, Barreto GE, Sahebkar A, Prevalence, pathological mechanisms, and genetic basis of limb-girdle muscular dystrophies: A reviewJ Cell Physiol 2019 234(6):7874-84.Epub 2018 Dec 710.1002/jcp.2790730536378 [Google Scholar] [CrossRef] [PubMed]
[4]. Mercuri E, Bushby K, Ricci E, Birchall D, Pane M, Kinali M, Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contracturesNeuromuscul Disord 2005 15:164-71.10.1016/j.nmd.2004.10.00815694138 [Google Scholar] [CrossRef] [PubMed]
[5]. Fanin M, Angelini C, Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: the yield and the pitfallsMuscle Nerve 2015 52:163-73.10.1002/mus.2468225900067 [Google Scholar] [CrossRef] [PubMed]
[6]. Beckmann JS, Spencer M, Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenanceNeuromuscul Disord 2008 18(12):913-21.10.1016/j.nmd.2008.08.00518974005 [Google Scholar] [CrossRef] [PubMed]
[7]. Khadilkar SV, Faldu HD, Patil SB, Singh R, Limb-girdle muscular dystrophies in India: A reviewAnn Indian Acad Neurol 2017 20(2):87-95.10.4103/aian.AIAN_81_1728615891 [Google Scholar] [CrossRef] [PubMed]
[8]. Pathak P, Sharma MC, Sarkar C, Jha P, Suri V, Mohd H, Limb girdle muscular dystrophy type 2A in India: A study based on semi-quantitative protein analysis, with clinical and histopathological correlationNeurol India 2010 58(4):549-54.10.4103/0028-3886.6867520739790 [Google Scholar] [CrossRef] [PubMed]
[9]. https://databases.lovd.nl/shared/genes/CAPN3 (last accessed on 20/02/2019) [Google Scholar]
[10]. Wang L, Zhang VW, Li S, Li H, Sun Y, Li J, The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patientsOrphanet Journal of Rare Diseases 2018 13(1):13310.1186/s13023-018-0859-630107846 [Google Scholar] [CrossRef] [PubMed]