Unilateral and Atypical Presentation of Vogt-Koyanagi-Harada Syndrome: A Case Report
Abhinav Dhami1, GS Dhami2
1 Consultant, Department of Cataract and Retina, Dhami Eye Hospital, Ludhiana, Punjab, India.
2 Medical Director, Department of Cataract and Refractive Surgery, Dhami Eye Hospital, Ludhiana, Punjab, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Abhinav Dhami, 82-B, Kitchlu Nagar, Ludhiana-141001, Punjab, India.
E-mail: drabhinavdhami@gmail.com
Vogt-Koyanagi-Harada (VKH) disease is a multisystem disorder with ocular manifestations including severe bilateral panuveitis with iridocyclitis, serous retinal detachment, diffuse choroidal swelling and optic disc hyperemia. We report a case of 41-year-old male who presented with diminution of vision in left eye with the presence of yellowish subretinal lesions throughout the posterior pole. There was no evidence of anterior or vitreous inflammation. The fundus fluoresce in angiography revealed early hypofluoroscent lesions with late phase showing multiple areas of pooling with no significant pin point hyperfluorescent lesions. The optical coherence tomography showed undulation of the outer retinal layers. Blood work up was within normal limits, Mantoux test showing 10×8 mm of induration. A differential diagnosis of atypical VKH disease versus central serous retinopathy was established. Oral steroids were started in close monitoring and marked resolution of the retinal lesions was noted. The case presents a unique scenario in which atypical VKH disease mimicked a central serous retinopathy like retinal picture and a correct diagnosis was established with the aid of Optical Coherence Tomograpy (OCT).
Optical coherence tomography, Uveitis, Uveomeningitic disease
Case Report
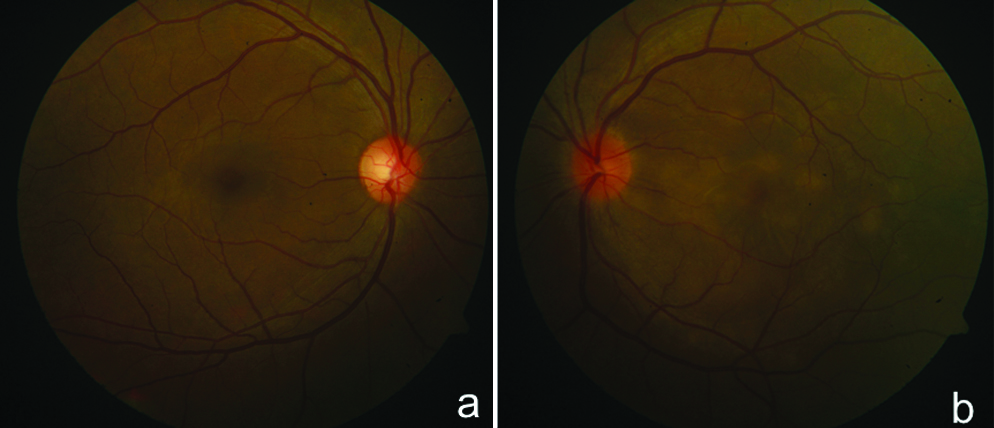
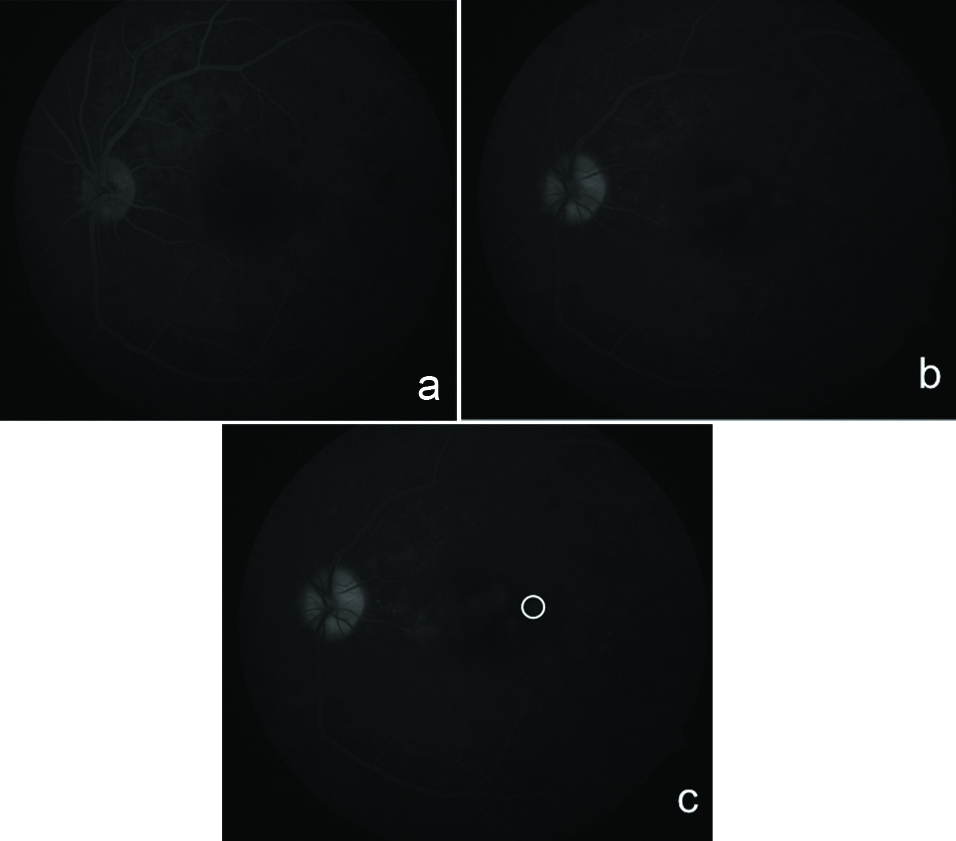
A 41-year-old male reported with decreased vision in left eye and visual acuity of 6/60 on Snellen’s chart and right eye was within normal limits [Table/Fig-1a]. On examination, there was no anterior chamber or vitreous reaction. The retinal evaluation showed mild blurring of disc margins with presence of multiple yellowish subretinal lesions throughout the posterior pole and internal limiting striae around the fovea [Table/Fig-1b]. The optical coherence tomography (OCT) showed multilobular undulation of the outer retinal layers with early serous retinal detachment from the disc upto to foveal center [Table/Fig-2a]. The fundus fluorescein angiography [Table/Fig-3a] showed early hypofluorescent lesions [Table/Fig-3b] around the posterior pole with late phase showed few pinpoint leaks (white circle) [Table/Fig-3c] with areas of pooling of dye.
(a) Fundus image of the right eye, (b) Fundus image of the left eye showing disc edema with presence of yellowish lesions throughout the posterior pole.
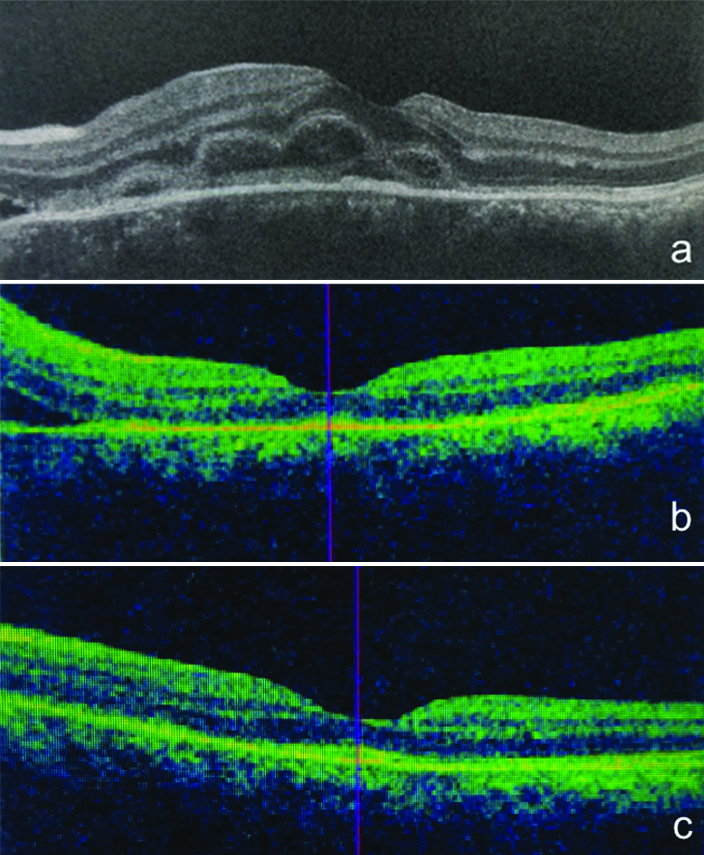
(a) Shows optical coherence tomography through the macula showing retinal thickening with multilobular undulation of the outer retinal layers with presence of sub retinal fluid nasally, (b) Showing resolution of the outer retinal layer undulation following treatment with oral steroids at one week and: (c) Shows complete resolution at one month.
(a) Shows FFA images of the left eye, (b) Shows early phase showing hypo-fluorescent lesions with leakage in the later frames, (c) Shows a white circle marking the areas of pinpoint leakage.
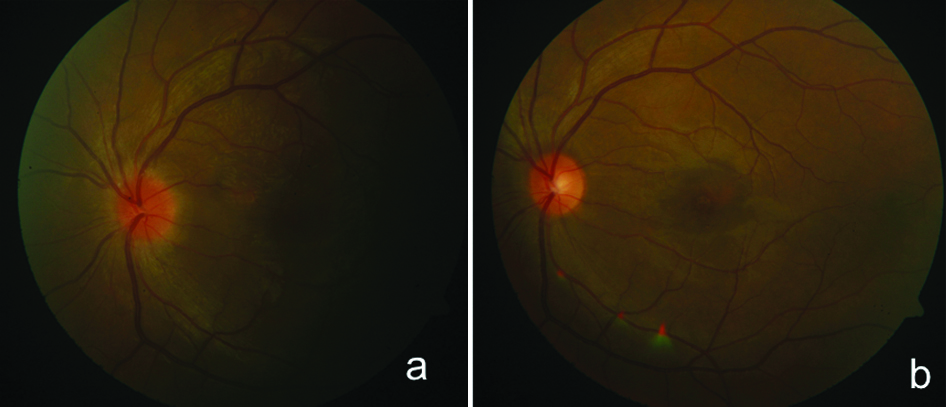
On systemic evaluation there was no evidence of associated complaints of tinnitus, poliosis or previous history of fever or meningitis. The B-scan ultrasonography revealed a retino-choroidal thickness of 1.2 mm in left eye. A differential diagnosis of atypical VKH and Central Serous Retinopathy (CSR) was made. In view of high suspicion of VKH syndrome oral steroids (prednisolone) were started on a close monitoring, at 80 mg/day with tapering dose of 10 mg/week, with oral ranitidine twice daily (150 mg) and oral Shelcal tablet (250 mg) twice daily. At one week follow-up, vision improved to 6/9 in left eye with complete resolution of the yellowish lesions with minimal disc swelling [Table/Fig-4a] and OCT showed complete resolution of edema at fovea [Table/Fig-2b]. At one month follow-up, vision had improved to 6/6 with complete resolution of disc edema [Table/Fig-2c and 4b] and was maintained up till last follow up (six months). The patient has been kept on low dose 5 mg oral prednisolone for preventing chances of recurrence and has maintained the visual recovery.
(a) Fundus image of left eye showing resolution of yellowish lesions and disc edema at one week of treatment, (b) Fundus image of left eye showing complete resolution.
Discussion
VKH is a multisystem autoimmune disorder and the diagnosis is established on the basis of bi-laterality, no history of penetrating ocular trauma or surgery, no other ocular disease, presence of auditory or neurological findings with typical integumentary changes [1-5]. Lin et al., analysed the findings between VKH and CSR using OCT and established that VKH presents with the presence of Retinal Pigment Epithelial (RPE) folds, fluctuation of the internal limiting membrane, presence of subretinal septa and undulations in retinal pigmented layer [4]. CSR was noted to present with pigment epithelial detachments and bulge of retinal pigment epithelium [4,6]. In this case, unilaterality with no inflammation deluded in reaching a correct diagnosis and the presence of multi-lobular outer retinal layer edema on OCT aided in the diagnosis [4]. Literature reports only a few atypical cases of VKH presenting either as CSR, choroiditis, neuro-retinitis and acute posterior multifocal placoid pigment epitheliopathy [4-6].
Conclusion
This case highlights the importance in differentiating VKH from a CSR, in eyes with unilateral involvement. It is important for clinician to consider VKH as a possibility in the absence of typical ocular and integumentary findings, even though it is a rare clinical variant of the disease and early treatment needs to be initiated, as delay in therapy can lead to irreversible visual loss.
[1]. Beniz JO, Forster DJ, Lean JS, Smith RE, Rao NA, Variations in clinical features of the Vogt-Koyanagi-Harada syndromeRetina (Philadelphia, Pa.) 1991 11(3):275-80.10.1097/00006982-199111030-000011961985 [Google Scholar] [CrossRef] [PubMed]
[2]. Ozdal P, Ozdamar Y, Yazici A, Teke MY, Ozturk F, Vogt-Koyanagi-Harada disease: Clinical and demographic characteristics of patients in a specialized eye hospital in TurkeyOcul Immunol Inflamm 2014 22(4):277-86.10.3109/09273948.2013.85644824328424 [Google Scholar] [CrossRef] [PubMed]
[3]. Baltmr A, Lightman S, Tomkins-Netzer O, Vogt–Koyanagi–Harada syndrome–current perspectives.Clin Ophthalmol (Auckland, NZ) 2016 10:2345-61.10.2147/OPTH.S9486627932857 [Google Scholar] [CrossRef] [PubMed]
[4]. Lin D, Chen W, Zhang G, Huang H, Zhou Z, Cen L, Comparison of the optical coherence tomographic characters between acute Vogt-Koyanagi-Harada disease and acute central serous chorioretinopathyBMC Ophthalmol 2014 14(1):8710.1186/1471-2415-14-8724974016 [Google Scholar] [CrossRef] [PubMed]
[5]. Agrawal A, Biswas J, Unilateral Vogt-Koyanagi-Harada disease: Report of two casesMiddle East Afr J Ophthalmol 2011 18(1):82-84.10.4103/0974-9233.7589821572744 [Google Scholar] [CrossRef] [PubMed]
[6]. Li B, Bentham RJ, Gonder JR, A case of unilateral and spontaneously resolving posterior uveitis with overlapping features of Vogt-Koyanagi-Harada disease and acute posterior multifocal placoid pigment epitheliopathySpringerplus 2016 5(1):147110.1186/s40064-016-3132-227652046 [Google Scholar] [CrossRef] [PubMed]