IgG4- Related TIN: Masquerading as Malignant Infiltrative Disorder
S Vijaya Devi1, Deepika Hemrajani2, Pankaj Beniwal3, Ranjana Solanki4
1 Senior Resident, Department of Pathology, SMS Medical College, Jaipur, Rajasthan, India.
2 Associate Professor, Department of Pathology, SMS Medical College, Jaipur, Rajasthan, India.
3 Associate Professor, Department of Nephrology, SMS Medical College, Jaipur, Rajasthan, India.
4 Professor, Department of Pathology, SMS Medical College, Jaipur, Rajasthan, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Ranjana Solanki, Professor, Department of Pathology, SMS Medical College, JLN Marg, Jaipur-302004, Rajasthan, India.
E-mail: solankiranjana@ymail.com
Immunoglobulin G4 Related Disease (IgG4-RD) is an autoimmune disease first described in pancreas. IgG4 RD has been reported in various organs and its renal involvement is known as IgG4-Related Kidney Disease (IgG4-RKD). Here, authors report a case of a 37-year-old male who presented with chronic kidney disease. He had generalized lymphadenopathy and hepatomegaly. Investigations revealed proteinuria, raised serum creatinine and high serum protein with marked hypergammaglobulinemia. Radiology revealed bilateral nephromegaly. Renal biopsy was done to rule the infiltrative disorder. Histopathological examination showed extensive periglomerular fibrosis, tubular atrophy and plasma cell-rich inflammatory infiltrate. Foci of storiform fibrosis were also seen. Serum IgG4 levels were found to be 49.3 g/L (reference range 0.03 to 2 g/L). Hence, a diagnosis of IgG4-TIN (Tubulointerstitialnephritis) was rendered. In a background of multiple organ involvement with characteristic histomorphological features a possibility of IgG4 RKD must always be considered.
Kidney disease, Malignancy, Pancreas
Case Report
A 37-year-old male presented with chief complaint of generalised weakness and bilateral leg swelling for three years. On clinical examination, he had cervical and axillary lymphadenopathy, mild hepatomegaly and pitting oedema. As per history, patient received Antitubercular Therapy (ATT) advised by local physician on basis of cough, dyspnea and chest X-ray findings six years back. however, his renal function deteriorated soon which improved with stopping ATT and supportive treatment at that time. No records of that event were available with the patient.
On lab investigation, his haemoglobin was 10.6 mg/dL and his counts were within normal range. His serum creatinine was high (3.6 mg/dL), serum total protein levels were elevated (12.01 g/dL) but serum albumin was low (2.7 g/dL) and transaminases were normal. Routine urine examination was within normal range. Hence, Serum Protein Electrophoresis (SPEP) was done, which showed marked hypergammaglobulinemia but no evident monoclonal gammopathy was noted. CT scan revealed nephromegaly with mildly increased renal cortical echogenicity. Peripheral blood film showed normocytic normochromic picture. Fine needle aspirate from cervical lymph node showed features of reactive lymphadenitis. In view of raised serum creatinine renal biopsy was done. Due to presence of multiple lymphadenopathy bone marrow biopsy was done to rule out presence of any infiltrative disorder.
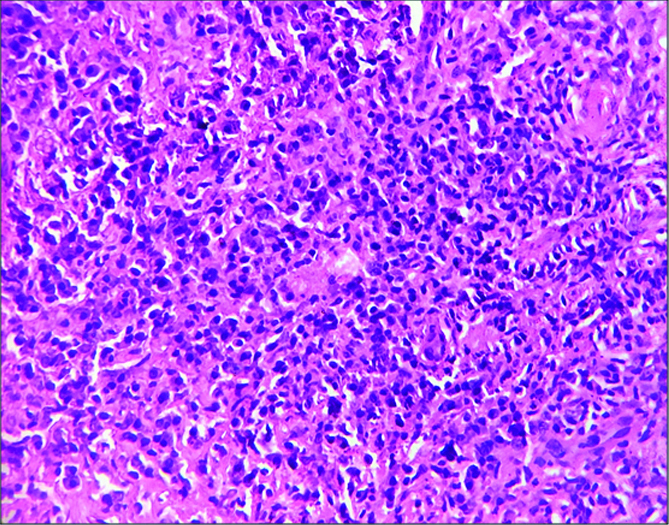
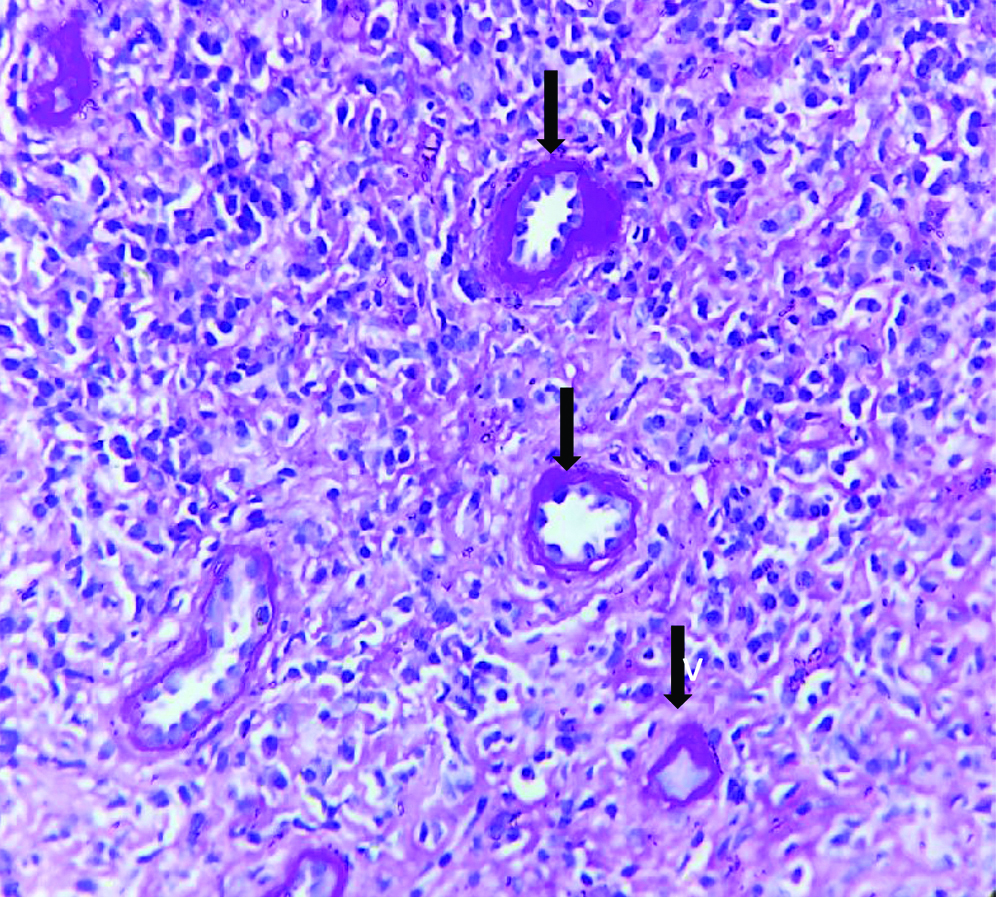
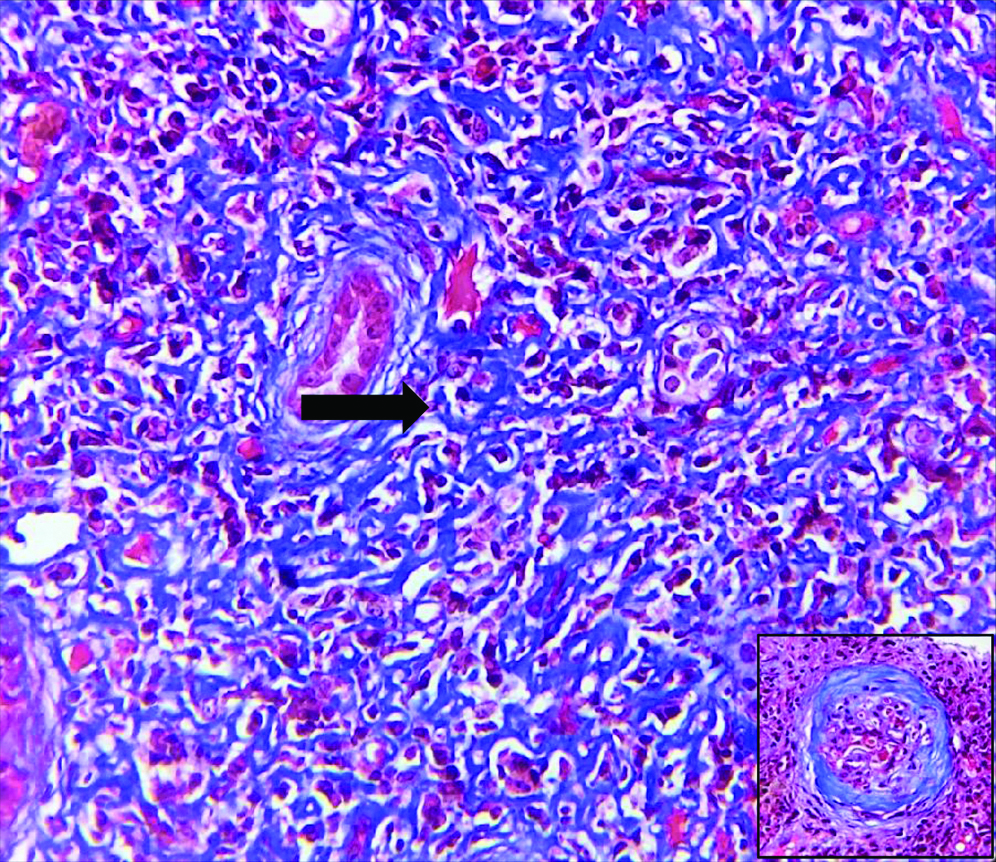
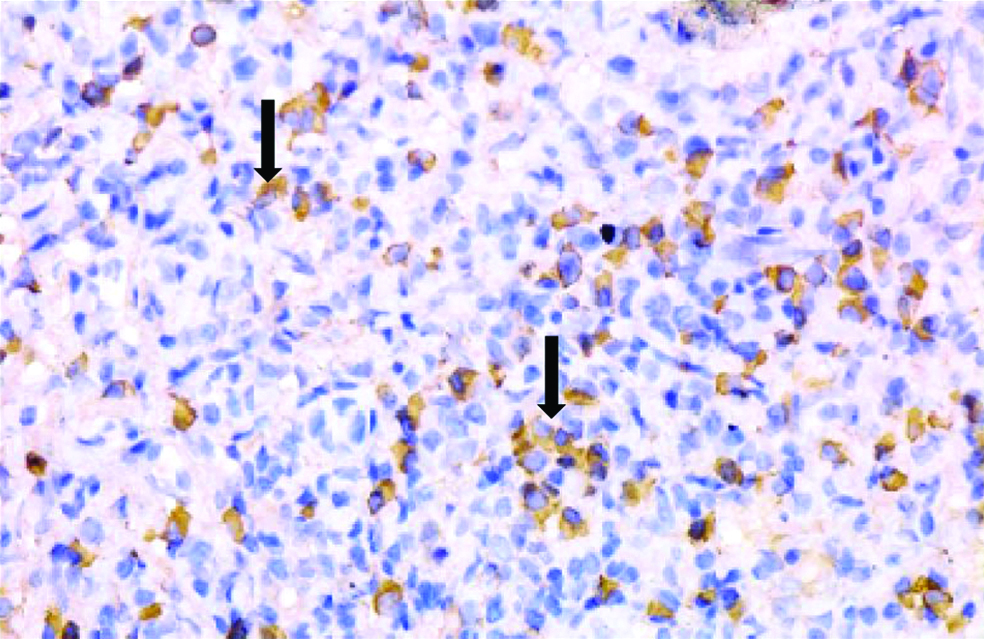
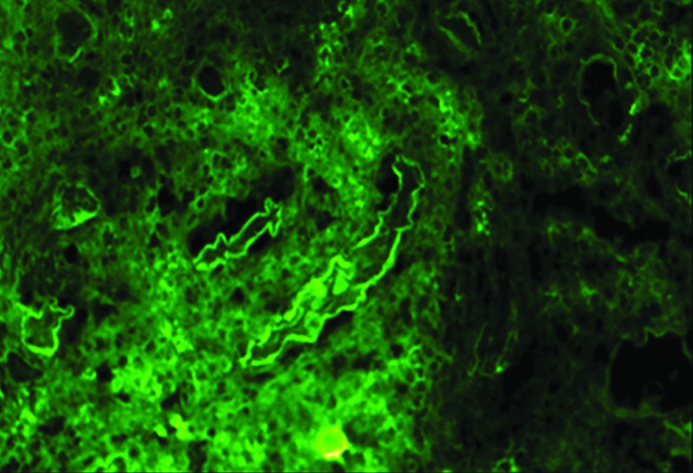
Histopathological examination of the kidney tru-cut biopsy revealed dense inflammatory infiltrate composed mainly of plasma cells and lymphocytes (H&E stain) [Table /Fig-1]. 40% of the glomeruli were obsolescent and some showed ischaemic shrinkage. Rest of the glomeruli showed mild mesangial expansion. Renal tubules showed thickening of tubular basement membrane (PAS Stain) [Table/Fig-2]. Extensive periglomerular fibrosis and foci of storiform fibrosis in interstitium were noted. (Masson Trichrome) [Table/Fig-3]. In view of characteristic histological features, a serum IgG4 level was subsequently done which was 49.3 g/L (ref range-0.03 to 2 g/L). Immunohistochemistry for IgG4 positive plasma cells revealed more than 10 IgG4+ plasma cells per HPF [Table/Fig-4]. No feature of membranous glomerulopathy was noted. Immunofluorescence (IF) microscopy revealed granular 1+ IgG deposition in TBM [Table/Fig-5]. He was treated with high dose corticosteroids (1 mg/kg/day prednisolone) to which he responded very well and later on maintenance dose of 5 mg/day. His serum creatinine levels have also returned to normal [1 mg/dL].
Inflammatory infiltrate composed mainly of plasma cells and lymphocytes (H&E 100X).
Interstitium showing renal tubular thickening (PAS x400).
Foci of storiform pattern in renal interstitium Inset: Periglomerular fibrosis (Masson Trichrome 100X).
Immunohistochemistry for IgG4 positive plasma cells revealed more than 10 IgG4+ plasma cells per HPF. (400x).
: Immunofluorescence (IF) microscopy revealed granular 1+ IgG deposition in tubular basement membrane.
Discussion
IgG4-RD is an autoimmune disease that often involves multiple organs, first recognised in pancreas [1]. This entity is a chronic immune mediated fibro-inflammatory condition characterised by infiltration of IgG4 positive plasma cells, fibrosis and obliterative phlebitis of the involved organs. IgG4-RD has been reported in almost every organ of the body including lung, liver, gall bladder, salivary and lacrimal gland, orbit, breast, retroperitoneum, aorta, lymph nodes, kidney, skin, pituitary gland and prostate. Renal involvement, known as IgG4 RKD, presents mainly as Tubulointerstitialnephritis (TIN) [2]. Membranous glomerulonephritis and obstructive nephropathy are other manifestations of IgG4-RKD [3]. However, varied the presentation may be, there are three histologic features that are characteristic for IgG4-RD. These include lymphoplasmacytic infiltrate rich in IgG4 plasma cells, storiform fibrosis and obliterative phlebitis [4]. This feature was not noted in the present case.
IgG4 RKD was first reported as a distinct entity in 2004 [5]. Four of the 35 patients of IgG4-RKD reported by Raissain Y et al., had bilateral markedly enlarged kidneys (≥14.5 cm) [6]. The most common CT scan finding was found to be multiple low-density lesions [7]. The present case had bilateral kidney enlargement with multiple areas of altered echotexture in renal parenchyma with maintained corticomedullary differentiation. CT scan is the most reliable technique in delineating the characteristics and distribution of the renal lesions [7], could not be done in the present patient because of renal impairment.
Increased serum IgG4 level is the most important serological finding in IgG4-RKD [7]. Raissain Y et al., reported 22 of 25 (88%) patients with increased total serum IgG or IgG4 or hypergammaglobulinemia in their study [6]. The present patient also had very high serum IgG4 levels of 49.30 g/L.
Characteristic histologic findings are one of the mandatory criteria for diagnosis of IgG4-RKD [Table/Fig-6] [6]. Some similarities in histological features may be seen in Rosai-Dorfman disease, Castleman disease, granulomatosis with polyangiitis and IgG4- lymphoma. Moreover, increased IgG4+ plasma cells can also be seen in necrotising glomerulonephritis, diabetic nephropathy, lupus nephritis, Rosai-Dorfman disease, Castleman disease, IgG4+ lymphoma, inflammatory myofibroblastic tumour, cutaneous plasmacytosis, inflammatory bowel disease and idiopathic TIN [8]. These differentials could be ruled out in the present case because of absence of vasculitis, granulomas, histiocytic proliferation, immature plasma cells and necrosis. The other important histopathological feature of IgG4-TIN - storiform fibrosis was seen at least focally in the biopsy [9,10], was present in the present case. Marked tubular atrophy and disappearance of tubules in some areas was also noted in the Haematoxylin and Eosin stained slides.
Diagnostic criteria for IgG4-TIN proposed by RaissianY et al., [6].
| Histology | Plasma cell-rich tublulointerstitial nephritis with >10 IgG4 + plasma cells/HPF in the most concentrated fielda Tubular basement membrane immune complex deposits by immunofluorescence, immunohistochemistry, and/or electron microscopyb
|
| Imaging | Small peripheral low-attenuation cortical nodules, round one wedge-shaped lesions, or diffuse patchy involvement Diffuse marked enlargement of kidneys
|
| Serology | Elevated serum IgG4 or total IgG level |
| Other organ involvement | Including autoimmune pancreatitis, sclerosis cholangitis, inflammatory masses in any organ, sialadenitis, inflammatory aortic aneurysm, lung involvement, and retroperitoneal fibrosis |
Diagnosis requires the histologic feature of plasma cell-rich TIN with increased IgG4+ plasma cells and at least one other feature from the categories of “imaging”, “Serology” or “other organ involvement”.
amandatory criterion; bsupportive criterion, present in >80% of cases
IgG and C3 deposition in TBM and interstitium has been reported in about 80% of IgG4 TIN by Immunofluorescence (IF) microscopy. Yamaguchi Y et al., analysed subclass expression in five cases, all of which showed IgG4 deposition in TBM and interstitium [10]. The present case showed only IgG deposition in TBM, IF could not be done for IgG4 subclass due to nonavailability of respective antibodies in the pathology laboratory.
Conclusion
IgG4-RKD is a rare disease and difficult to diagnose as it has varied clinical and imaging findings. IgG4-TIN, MGN and obstructive nephropathy are three main manifestations. Although renal biopsy is important in diagnosis of IgG4 RKD, no single test is confirmatory. A combination of clinical, radiological, histological and serological findings is required for the diagnosis. The main stay of treatment is corticosteroids. Serum IgG4 levels rapidly decrease after corticosteroid therapy. Immunosuppressants are considered in refractory or recurrent cases.
Diagnosis requires the histologic feature of plasma cell-rich TIN with increased IgG4+ plasma cells and at least one other feature from the categories of “imaging”, “Serology” or “other organ involvement”.
amandatory criterion;
bsupportive criterion, present in >80% of cases
[1]. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, High serum IgG4 concentrations in patients with sclerosing pancreatitisN Engl J Med 2001 344(10):732-38.10.1056/NEJM20010308344100511236777 [Google Scholar] [CrossRef] [PubMed]
[2]. Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritisKidney International 2010 78(10):1016-23.10.1038/ki.2010.27120720530 [Google Scholar] [CrossRef] [PubMed]
[3]. Watson SJW, Jenkins DAS, Bellamy COS, Nephropathy in IgG4-related Systemic DiseaseAm J SurgPathol 2006 30(11):1472-77.10.1097/01.pas.0000213308.43929.9717063091 [Google Scholar] [CrossRef] [PubMed]
[4]. Zheng K, Teng F, Li XM, Immunoglobulin G4 related kidney disease: Pathogenesis, diagnosis, and treatmentChronic Dis Transl Med 2017 3(3):138-47.10.1016/j.cdtm.2017.05.00329063068 [Google Scholar] [CrossRef] [PubMed]
[5]. Takeda Si, IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitisNephrol Dial Transplant 2004 19(2):474-76.10.1093/ndt/gfg47714736977 [Google Scholar] [CrossRef] [PubMed]
[6]. Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, Diagnosis of IgG4-related tubulointerstitial nephritisJ Am SocNephrol 2011 22(7):1343-52.10.1681/ASN.201101006221719792 [Google Scholar] [CrossRef] [PubMed]
[7]. Kawano M, Saeki T, IgG4-related kidney disease-an updateCurr Opin Nephrol Hypertens 2015 24(2):193-201.10.1097/MNH.000000000000010225594543 [Google Scholar] [CrossRef] [PubMed]
[8]. Deshpande V, The pathology of IgG4-related disease: critical issues and challengesSemin Diagn Pathol 2012 29(4):191-96.10.1053/j.semdp.2012.08.00123068297 [Google Scholar] [CrossRef] [PubMed]
[9]. Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, A new clinicopathological entity of IgG4-related autoimmune diseaseJ Gastroenterol 2003 38(10):982-84.10.1007/s00535-003-1175-y14614606 [Google Scholar] [CrossRef] [PubMed]
[10]. Yamaguchi Y, Kanetsuna Y, Honda K, Yamanaka N, Kawano M, Nagata M, Characteristic tubulointerstitial nephritis in IgG4-related diseaseHum Pathol 2012 43(4):536-49.10.1016/j.humpath.2011.06.00221889187 [Google Scholar] [CrossRef] [PubMed]