Primary Ewing’s Sarcoma of Adrenal Gland-A Rare Case
Krishnendu Maity1, Akash Agrawal2, Chhanda Datta3, Dilip Kumar Pal4
1 Assistant Professor, Department of Urology, Institute of Post Graduate Medical Education and Research, Kolkata, West Bengal, India.
2 Postdoctoral Trainee, Department of Urology, Institute of Post Graduate Medical Education and Research, Kolkata, West Bengal, India.
3 Professor and Head, Department of Pathology, Institute of Post Graduate Medical Education and Research, Kolkata, West Bengal, India.
4 Professor and Head, Department of Urology, Institute of Post Graduate Medical Education and Research, Kolkata, West Bengal, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Dilip Kumar Pal, 244, AJC Bose Road, Kolkata-700020, West Bengal, India.
E-mail: urologyipgmer@gmail.com
Ewing’s Sarcoma (ES) or Primitive Neuro-Ectodermal Tumour (PNET) typically occurs in long or flat bones, the chest wall, extra-skeletal soft tissue, and rarely in solid organs. Incidence of urological ES is uncommon. Here, we report a case of primary ES of the right adrenal gland in a 16-year-old boy who presented with an adrenal mass, with a successful outcome.
Mesenchymal adrenal mass, Rare adrenal tumour, Sarcoma
Case Report
A 16-year-old Hindu male patient presented in outpatient clinic with eight months history of dull aching, continuous, non radiating, right sided upper abdominal pain, that was relieved by analgesics. There was no history of fever, vomiting, haematuria, graveluria, malaise, weight loss. There was also no history of haemoptysis, diaphoresis or back pain.
General and systemic examinations were normal. Routine blood and urine investigations were normal. Ultrasonography revealed a mass in right suprarenal region. On Contrast enhanced CT, single, large, heterogenous enhancing mass lesion measuring 10.4×9.0×7.4 cm in right suprarenal region, with areas of necrosis and internal vascularity, was noted. There was partial encasement of IVC and fat planes of upper pole of kidney was involved [Table/Fig-1]. Functional study of the adrenal gland was negative.
Pre chemotherapy CECT showing large right adrenal mass.
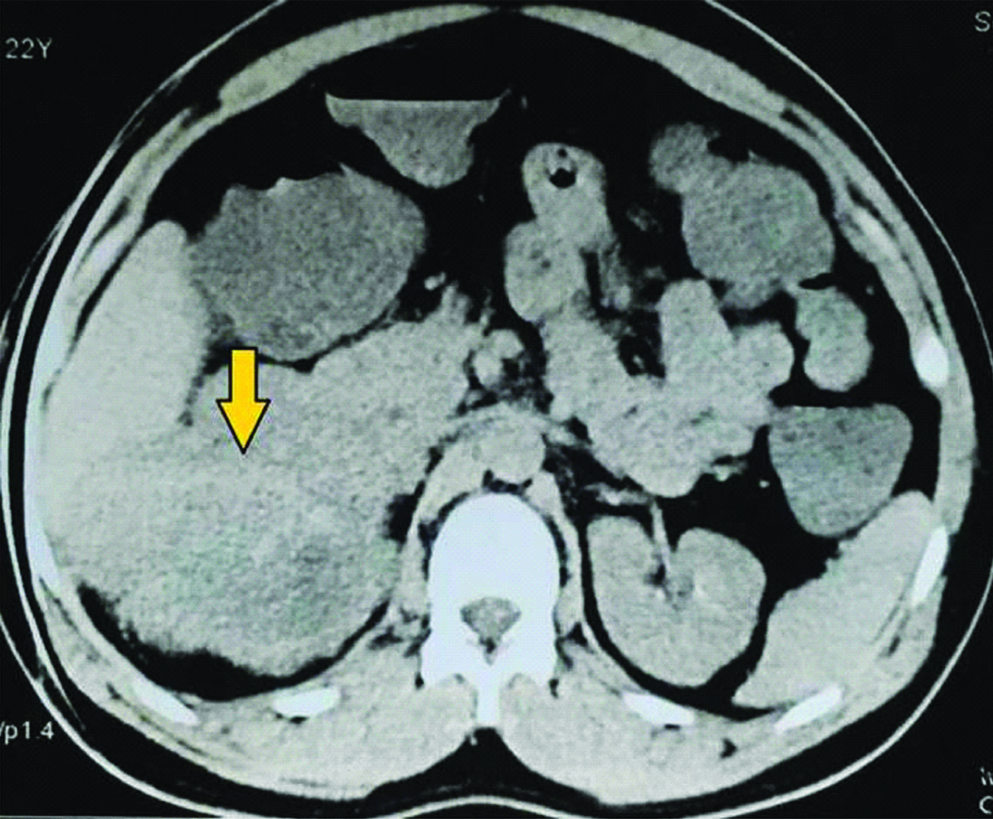
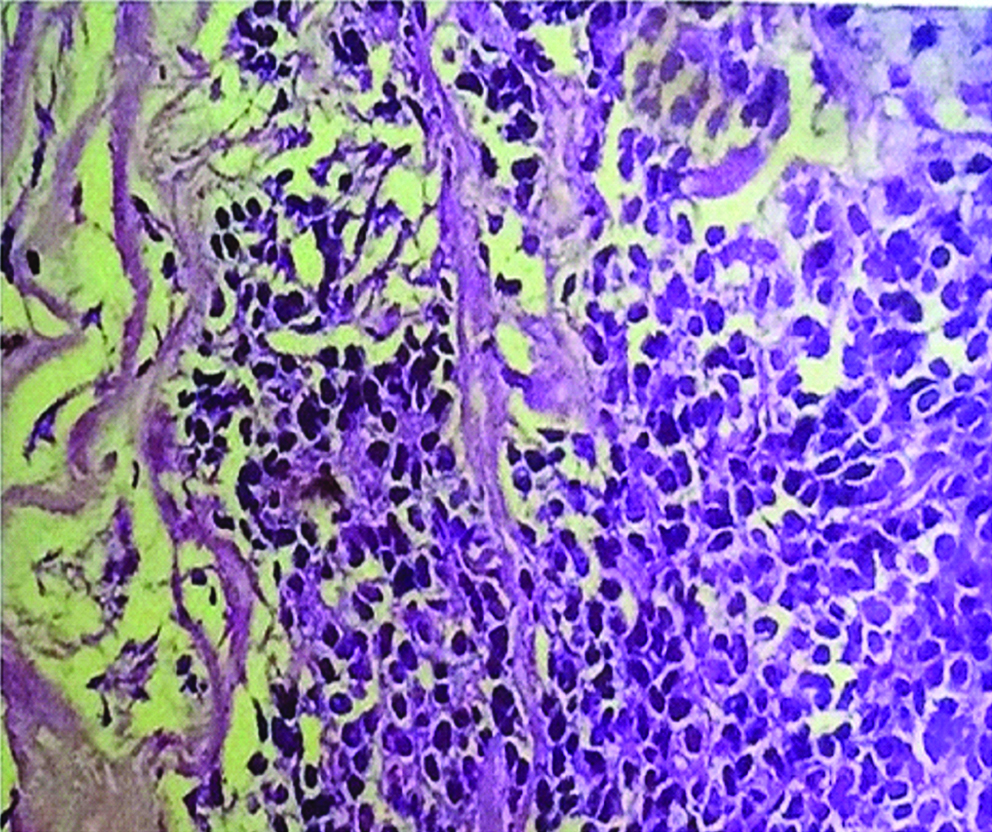
At that stage, imaging features gave the sense of inoperability. Core needle biopsy revealed malignant small round cell tumour; suggestive of ewing’s sarcoma [Table/Fig-2]. Chemotherapy was planned and vincristine, ifosfamide, vincristne and doxorubicin based six cycles of chemotherapy was given over a period of four months.
Core biopsy (40x) HPE showing small round cells.
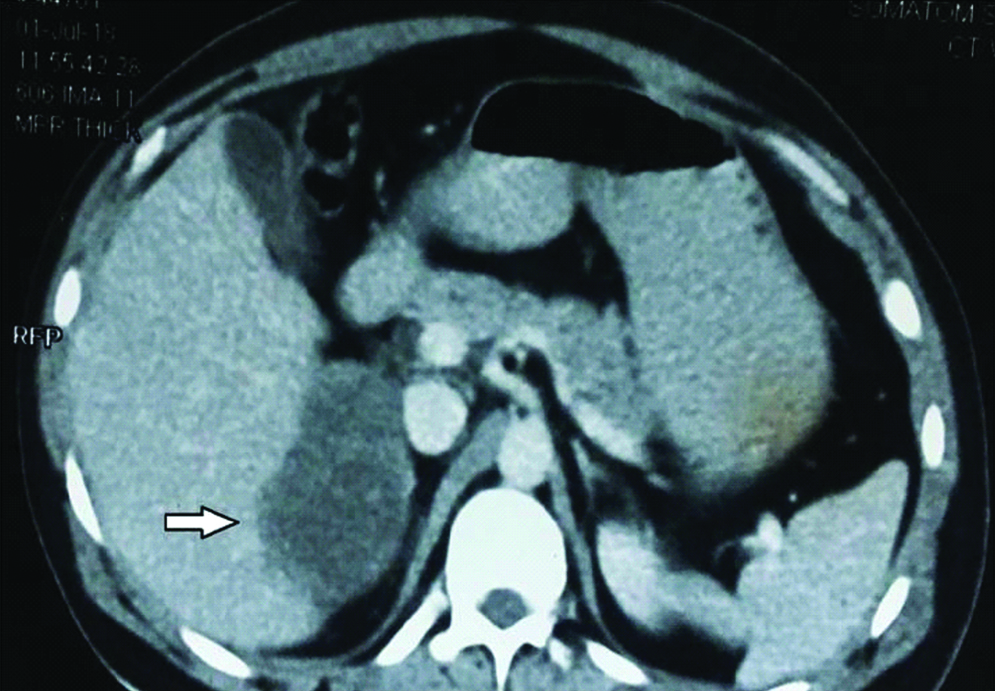
Repeat contrast enhanced CT was done which revealed significant reduction in size of mass to 8.1×6.9×6.5 cm [Table/Fig-3]. Right adrenalectomy with consent for right nephrectomy was planned, and with all preoperative preparation and optimization patient was taken for surgery. Right sided subcostal incision was given extending to left subcostal region in chevron fashion, and abdomen was entered intraperitoneally. There was no liver metastasis. Intraoperatively, adrenal was not dissectible from upper pole of right kidney and the hilar vessels were stretched over the tumour, so right adrenalectomy along with right radical nephrectomy was performed along with excision of hilar lymph nodes. As mass was not separable from inferior border of the liver, part of inferior border was also resected along with the mass. Enmass resection was done [Table/Fig-4].
Post chemotherapy CECT showing decrease in size of adrenal mass.
Resected mass with inferior border of liver and right kidney.
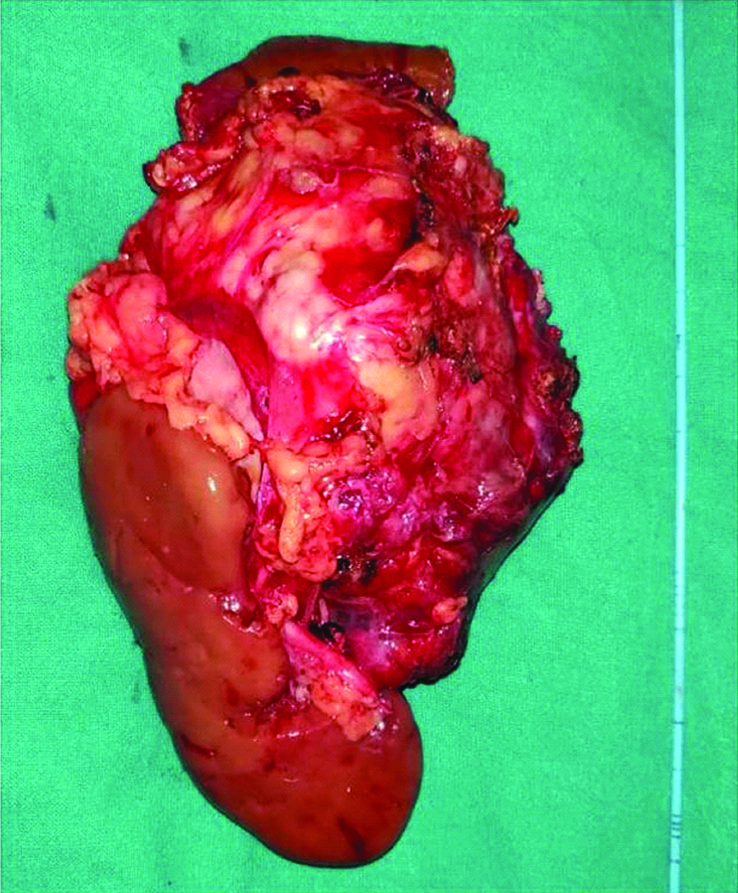
Postoperative course was uneventful except there was mild bilious outcome from drain for 5 days, which subsided. Patient was discharged on 8th postoperative day. On gross examination tumour was of 8x6x5 cm identified involving the right adrenal gland, whose cut surface was variegated and macroscopic extension was limited to the right adrenal. On histological examination tumour was composed of small round cells with rounded nuclei and scanty cytoplasm, the cells were arranged in sheets and cohesive lobules and showed rosettes and perivascular pseudorosettes. All the margins including gerotas fascia and renal vein were uninvolved. Lymphovascular invasion was present. On immunohistochemistry analysis CD99 was strongly positive [Table/Fig-5], synaptohysin was positive [Table/Fig-6], and Leukocyte Common Antigen was negative in tumour cells. At six months of follow-up, patient is doing well.
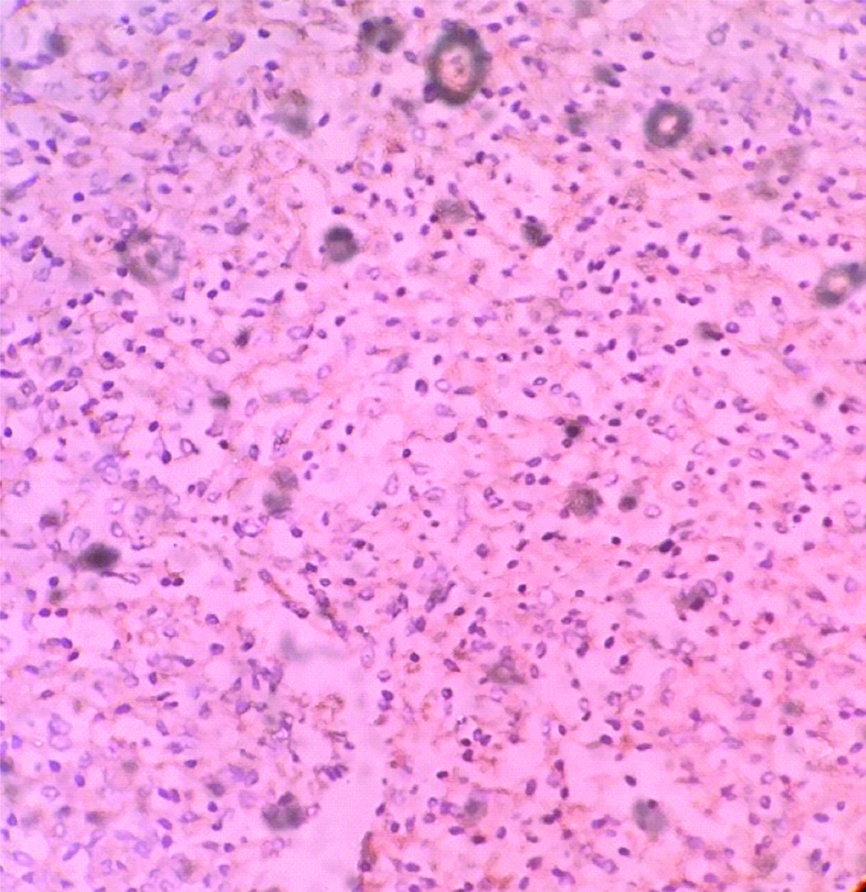
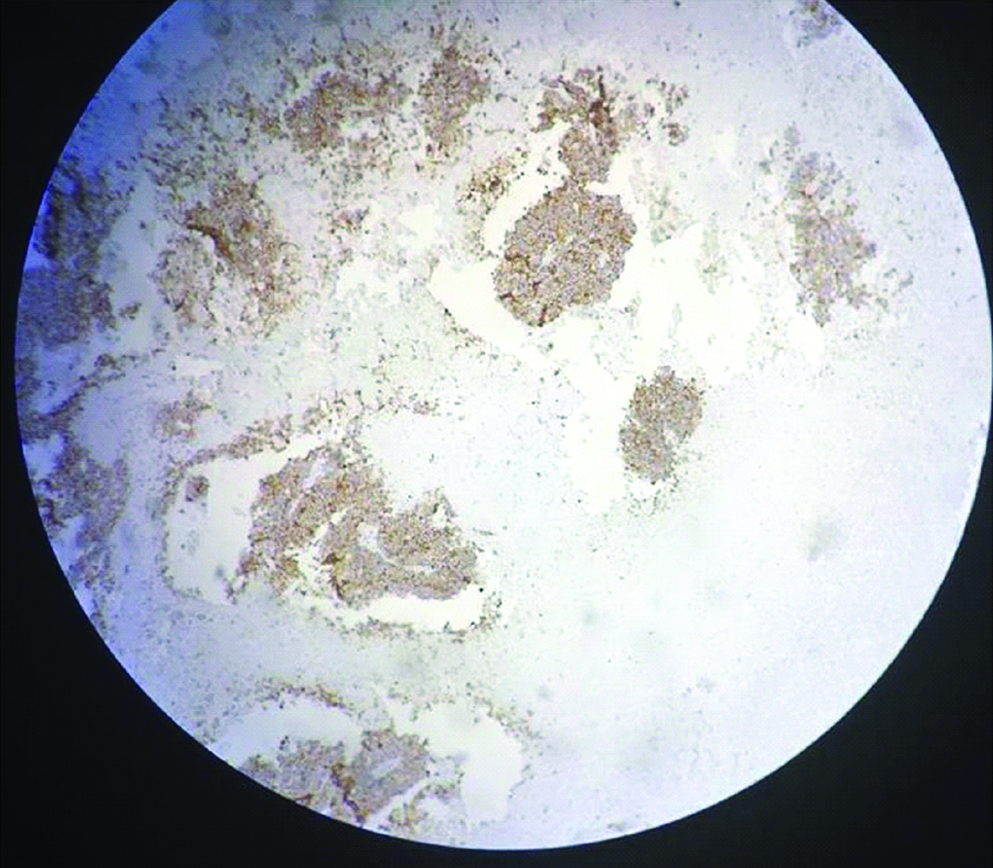
Discussion
Primary ES of adrenal gland is a very rare tumour [1]. Its first case was described by Marina et al., in 1989. Since then almost 35 cases have been reported. ES occurs mostly in 2nd decade with slight male predominance. Patients usually present with flank pain and rapidly growing mass [2]. Ultrasonography can be used as a screening tool but CECT is essential to find out the relation with the surrounding structures. CECT imaging helps to distinguish between benign and malignant adrenal tumours through its contrast washout characteristics [3]. Hormonal studies should be done to differentiate it from the functional adrenal tumours. It is difficult to make a definitive diagnosis of ES even on histopathology without Immunonohistochemistry [4]. CD 99 is a highly sensitive marker for ES. Patients show reciprocal translocation of t (11;22) (q24;q12) involving the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 in more than 90% of cases but this is not routinely assessed [5].
These are very rapidly growing and aggressive tumours and surgical resection is the mainstay of its treatment [6]. As this is a radio and chemo sensitive tumour so adjuvant chemotherapy and radiotherapy are advocated for better disease control but there is no consensus on standard chemo-radiation regimen [7]. Eddaoualline H et al., in their study also gave their patient neoadjuvant chemotherapy which was responded well as in our case [2]. Proper postoperative surveillance with imaging modalities and adjuvant chemotherapy might alter the outcome in individual cases. Some studies have demonstrated worse prognosis for adults compared with their younger counterparts [8,9].
Conclusion
Ewing’s sarcoma/PNET of the adrenal gland is extremely rare. One should always consider this rare diagnosis when a young patient presents with a large non-functional adrenal mass, which cannot be differentiated on imaging. Imaging studies along with biopsy is the proper way to proceed towards this rare diagnosis. Biopsy should be done in cases with larger nonfunctional masses/ inoperable tumours/equivocal imaging findings/unknown nature of tumour.
[1]. Tsang Y, Lang B, Tam S, Wong K, Primitive neuroectodermal adrenal gland tumourHong Kong Medical Journal 2014 20(5):444-46.10.12809/hkmj13412725307073 [Google Scholar] [CrossRef] [PubMed]
[2]. Eddaoualline H, Mazouz K, Rafiq B, EL Mghari Tabib G, EL Ansari N, Belbaraka R, Ewing sarcoma of the adrenal gland: A case report and review of the literatureJournal of Medical Case Reports 2018 12(1):01-05.10.1186/s13256-018-1601-729544549 [Google Scholar] [CrossRef] [PubMed]
[3]. Dunnick N, Korobkin M, Francis I, Adrenal radiology: Distinguishing benign from malignant adrenal massesAmerican Journal of Roentgenology 1996 167(4):861-67.10.2214/ajr.167.4.88193728819372 [Google Scholar] [CrossRef] [PubMed]
[4]. Zhang Y, Li H, Primitive neuroectodermal tumours of adrenal glandJapanese Journal of Clinical Oncology 2010 40(8):800-04.10.1093/jjco/hyq05020430773 [Google Scholar] [CrossRef] [PubMed]
[5]. Quezado M, Benjamin D, Tsokos M, EWS/FLI-1 fusion transcripts in three peripheral primitive neuroectodermal tumours of the kidneyHuman Pathology 1997 28(7):767-71.10.1016/S0046-8177(97)90147-7 [Google Scholar] [CrossRef]
[6]. Komatsu S, Watanabe R, Naito M, Mizusawa T, Obara K, Nishiyama T, Primitive neuroectodermal tumour of the adrenal glandInt J Urol 2006 13:606-07.10.1111/j.1442-2042.2006.01361.x16771733 [Google Scholar] [CrossRef] [PubMed]
[7]. Malpica A, Moran C, Primitive neuroectodermal tumour of the cervix: A clinicopathologic and immunohistochemical study of two casesAnnals of Diagnostic Pathology 2002 6(5):281-87.10.1053/adpa.2002.3573912376920 [Google Scholar] [CrossRef] [PubMed]
[8]. Nesbit ME Jr, Gehan EA, Burgert EO Jr, Vietti TJ, Cangir A, Tefft M, Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup studyJ Clin Oncol 1990 8(10):1664-74.10.1200/JCO.1990.8.10.16642213103 [Google Scholar] [CrossRef] [PubMed]
[9]. Cangir A, Vietti T, Gehan E, Burgert E, Thomas P, Tefft M, Ewing’s sarcoma metastatic at diagnosis results and comparisons of two intergroup Ewing’s sarcoma studiesCancer 1990 66(5):887-93.10.1002/1097-0142(19900901)66:5<887::AID-CNCR2820660513>3.0.CO;2-R [Google Scholar] [CrossRef]