Unusual Low-grade Occipito-temporal Ganglioglioma with Calcification
AK Kapoor1, Divya Saxena2
1 Pathologist, Department of Pathology, RML Mehrotra Pathology Pvt. Ltd., Nirala Nagar, Lucknow, India.
2 Pathologist, Department of Pathology, RML Mehrotra Pathology Pvt. Ltd., Nirala Nagar, Lucknow, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: AK Kapoor, D87, Mahanagar Extension, Nirala Nagar-226020, Lucknow, India.
E-mail: drashokkapoor2016@gmail.com
Ganglioglioma (GG) is a rare benign tumour which may present as focal convulsions. Complete resection of the tumour may lead to excellent recovery. The present case report describes the features of a mixed neuronal-glial tumour in a young female patient. She complained of seizures for the last several years. Magnetic Resonance Imaging (MRI) was done. T1-weighted MRI revealed a hypointense cystic lesion in right occipitotemporal region. It measured 2.8×2.2×2.2 cm. The t2-weighted image showed a hyperintense mural nodule which measured 0.4 cm approximately. The craniotomy was done and the tumour was resected. Microscopically, the tumour showed the proliferation of astrocytes and dysplastic multinuclear neuronal cells. Dysplastic cells were large with round nuclei, abnormal clustering and irregular distribution of variably sized neurons. Few multinuclear neurons were also seen. Neoplastic neuronal cells were both synaptophysin and chromogranin A positive. Proliferated astrocytes showed pilocytic features. Eosinophilic granular bodies and perivascular inflammation were also seen. The tumour was diagnosed as a case of the World Health Organisation (WHO) Grade I ganglioglioma.
Benign neoplasm, Dysplastic neuronal cells, Proliferated astrocytes, Seizures
Case Report
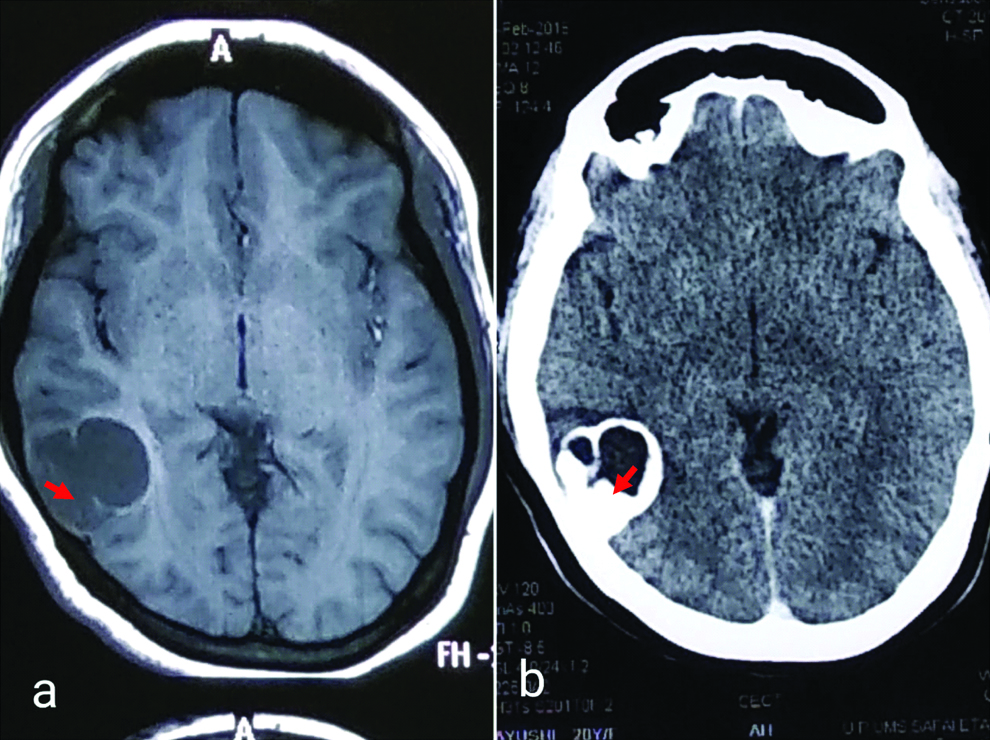
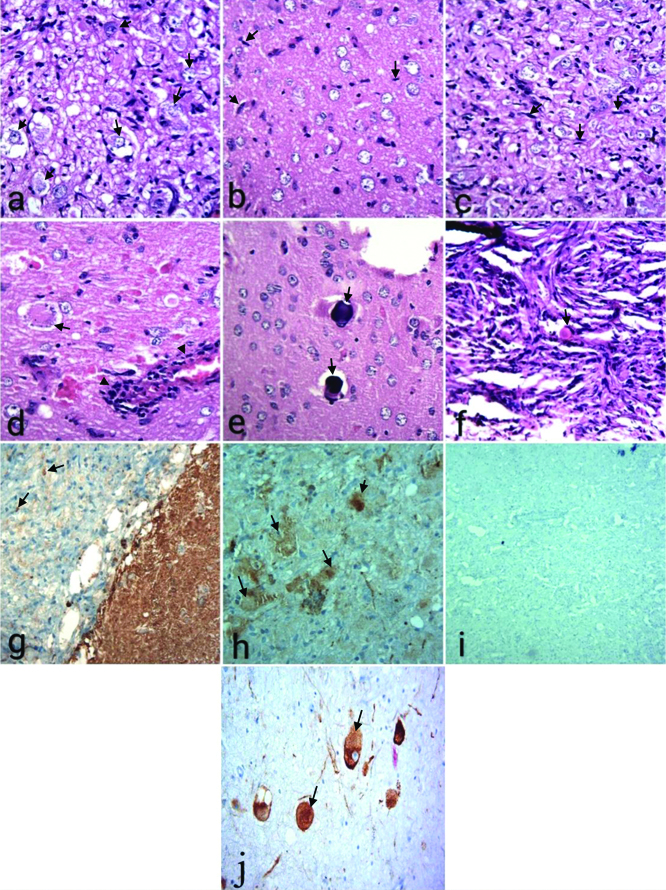
A 21-year-old female patient presented with seizures, headache and vertigo for the last eight years. The left-sided focal seizure was preceded by a headache, vertigo and confusion. The patient did not give the history of Tuberculosis (TB) in the family. On radiological examination, T1-weighted MRI showed a hypointense cystic mass in the occipitotemporal region. It measured 2.8×2.2×2.2 cm [Table/Fig-1a]. The T2-weighted image showed a hyperintense mural contrast-enhancing solid mass, measuring 0.4 cm in diameter [Table/Fig-1b]. The lesion was cortically-based. The patient was provisionally diagnosed as a case of glioma. The posterior right craniotomy was done and several small bits of tissue were collected during operation. Later, the specimen consisting of tissue bits was sent for histopathological examination. Microscopic examination showed neoplastic tumour mass. It consisted of dysplastic neuronal cells and proliferated glial cells. Dysplastic neuronal cells were large with round vesicular nuclei having prominent nucleoli. Few multinuclear neuronal cells were also seen [Table/Fig-2a]. Another bit microscopically showed a large number of pilocytic astrocytes along with Rosenthal fibres [Table/Fig-2b]. Astrocytic tumour cells also formed fascicles with elongated nuclei [Table/Fig-2c]. Surrounding peritumoral brain tissue showed neurons with the peripheral rim of Nissl granules near the plasma membrane (chromatolysis). Perivascular lymphocytic infiltration (pseudoencephalitis) was also seen [Table/Fig-2d]. Multiple focal areas of metastatic calcification (calcospherules or Psammoma bodies) were appreciated [Table/Fig-2e]. Eosinophilic granular bodies of lysosomal derivation were seen [Table/Fig-2f]. On the basis of histopathological findings, she was diagnosed as a case of Astrocytoma or Oligodendroglioma. Immunohistochemistry (IHC) showed positivity for Glial Fibrillary Acidic Protein (GFAP) of pilocytic astrocytes and Rosenthal fibres [Table/Fig-2g]. Surface cytoplasmic staining of neoplastic neuronal cells for synaptophysin was seen [Table/Fig-2h]. Additionally, intense diffuse cytoplasmic staining of neoplastic neuronal cells for chromogranin A was obtained. Rare mitoses and negative staining of most of the tumour cells for anti Ki-67 suggested a low proliferative index (3%) of the current tumour [Table/Fig-2i]. Anti-chromogranin A showed positive cytoplasmic staining of tumour cells [Table/Fig-2j]. Pathological and molecular findings were suggestive of a WHO Grade I GG. The patient was followed for three weeks after craniotomy and during this period she did not develop seizures. No other treatment was given. The patient appeared to have been cured of headache, vertigo and convulsions suggesting an excellent recovery.
a) T1-weighted MRI showed a hypointense cystic lesion in occipito-temporal area. Lesion measured 2.8×2.2×2.2 cm approximately; b) MRI (flair) showed an isointense lesion with hyperintense margin. Lesion contained a nodular tumour, measuring 0.4 cm in diameter. (←) shows tumour tissue.
a) Neuronal tumour cells showed mild nucleomegaly (↓) and mild pleomorphism (HE × 400); b) Tumour cells showed proliferated astrocyes (H&E ×400); c) Astrocytic tumour cells formed fascicles with elongated (↓) nuclei (H&E ×400); d) Surrounding brain parenchyma showed chromatolysis (↓). Perivascular chronic inflammation (↓) was also seen (H&E ×100); e) Psammoma body (↓) was seen (H&E × 100); f) Eosinophilic globular hyaline body (↓) was also seen (H&E ×400); g) Immunohistochemical (IHC) examination using anti-Glial Fibrillary Acidic Protein (GFAP) antibody showed mild staining for astrocytic glial (↓) processes (H&E ×100); h) IHC using anti-synaptophysin antibody showed positive cytoplasmic staining (↓) of neuronal tumour cells (×100); i) Anti Ki-67 antibody did not stain most of tumour cells, suggesting a low proliferative potential (×100); j) IHC showed positive cytoplasmic staining (↓) of neuronal tumour cells with anti-chromogranin A antibody (×400).
Discussion
GGs are rare intra-axial tumours. For example, GGs were diagnosed in 5 of 1560 brain tumours in a hospital-based study [1]. In another study, 156 cases of GGs (adults 115 and children 41) were reported in 30-years [2]. Present tumour was situated in occipitotemporal region. Similar tumours are relatively more common in temporal lobe when compared to other regions [2,3]. Moreover, GGs in other areas may have a less favourable clinical course than those in temporal lobe [4]. Earlier, an anaplastic posterior fossa infratentorial GG was reported [5]. Another case of GG at the cerebellopontine angle has been recorded [6]. Rarely, GG may present as a cerebellar tumour in paediatric age-group [7]. Another patient with tumour in parieto-occipital region has been reported [8]. Clinically, the previous patient presented with headache, confusion and slurred speech [8]. Cranial nerve cases are also on record. Clinically, the current tumour presented with focal seizures for the last eight years. Vestibular epilepsy and vertigo are rare and are intermixed with epileptic manifestations. Similar observations were reported in a previous study [3]. In another study, age at the onset of seizures was seven years or less in most (73%) of GG patients [9]. Most of GGs are amicable to complete surgical excision and are associated with excellent recovery [2]. Individual cases of GG have occurred in association with a family history of neurofibromatosis and Peutz-Jeghers syndrome. A candidate for the diagnosis of GG should not have LDH1 or LDH2 mutations which may be associated with infiltrative Astrocytoma and Oligodendroglioma [10]. In addition, a subset of ganglioglioma is known to harbor p.V600E mutation in BRAF oncogene [11]. Loss of p19 expression and p53 mutation may be involved in the malignant transformation of ganglioglioma [12]. The most interesting feature of the current case was the detection of a mural nodule in right occipitotemporal region. The nodule was located inside a cyst. Microscopically, the nodule consisted of tumour tissue. It showed proliferated astrocytes and dysplastic neuronal cells, suggesting it to be a mixed neuronal-glial cell tumour. Dysplastic cells were large with a dysmorphic neuronal appearance different from those of cortical neurons. Neoplastic neuronal cells also exhibited cytoplasmic staining for synaptophysin and chromogranin A. Conversely, non-neoplastic neurons did not stain for these markers. The neuronal component may also express CD34. Dysplastic neural cells of this type with intratumoural CD34 immunoreactivity is against infiltrating astrocytoma are oligodendroglioma. A patient with a cerebral mural nodule inside a cyst should be differentiated between a ganglion cell tumour, pilocytic astrocytoma or pleomorphic xanthoastrocytoma. Later lesion histologically may appear malignant yet its biological behaviour may be favourable. Moreover, staining for epithelial membrane antigen may be negative [13]. GGs are classified as WHO Grade I or II. Anaplasia or necrosis in the glial component may increase the WHO Grade to III or IV [14]. Current tumour had proliferated pilocytic astrocytes without anaplasia. Proliferation was believed to be limited to the glial component [15]. Rare mitoses and poor proliferative Ki-67 index of current tumour suggested it have a low proliferative potential. GGs are rare primary, slow-growing benign tumours. However, recurrence and malignant change may occur in a few patients [16]. Risk of malignant transformation is related to glial features [2]. Using a miRNA assay; miR-519d and miR-4758 were found to be upregulated in GG when compared to control cortex, peritumoral tissue, Dysembryoplastic neuroepithelial tumour, Astrocytoma and Glioblastoma [17]. Present case was unusual; about 11 cases of GG have been reported earlier from India [ 1, 6, 12 , 13, 18 ]. Second , neoplasm contains both neuronal cells and glial cells , suggesting its biphasic character. Third, perivascular lymphocytic infiltration , chromatolysis and presence of psammoma bodies further suggested the diagnosis of GG. Fourth, GFAP positivity of astrocytes; cytoplasmic staining of neoplastic neuronal cells for synaptophysin and chromogranin A favoured it to be GG. Fifth, anti-Ki-67 did not stain most of tumor cells, suggesting low proliferation index of current neoplasm.
Conclusion
Ganglioglioma is a benign biphasic neoplasm which consists of proliferated glial and dysplastic neuronal cells. It may be associated with intestinal hamartomas and mucocutaneous melanin pigmentation. Rarely, tumour recurrence and malignant change may develop. Several factors, e.g., frequent mitoses, high Ki-67 index (>45%), anaplastic glial components involving one-third of tumour bulk, the subcortical intraventricular location may indicate more aggressive behaviour. In addition, subtotal resection of tumour may result in recurrence and/or CSF-borne metastasis which may prove fatal if no treatment is given.
[1]. Nair V, Suri VS, Tatke M, Saran RK, Malhotra V, Singh D, Gangliogliomas: A report of five casesInd J Cancer 2004 41:41-46. [Google Scholar]
[2]. Zieman G, Dardis C, Gomes A, Scheck A, Eschbacher J, Ashby L, Predicting progressive gangliogliomas: Early identification of those at risk in a sample of 156 consecutive patients treated at the Barrow neurological institute (S22.004)Neurology 2014 82(10 supplement)April 29 [Google Scholar]
[3]. Ildan F, Tuna M, Gocer IA, Erman T, Cetinalp E, Intracerebral ganglioglioma: Clinical and radiological study of eleven surgically treated cases with follow-upNeurosurg Rev 2001 24(2-3):114-18.10.1007/PL0001239311485231 [Google Scholar] [CrossRef] [PubMed]
[4]. Patibandla MR, Ridder T, Atypical paediatric ganglioglioma is common and associated with a less favourable clinical courseJ Neurosurg paediat 2016 17:41-48.10.3171/2015.6.PEDS1521526431248 [Google Scholar] [CrossRef] [PubMed]
[5]. Bouali S, Maatar N, Zehani A, Mahmoud M, Kallel J, Jemel H, A case of adult anaplastic cerebellar gangliogliomaSurg Neurol Int 2018 9:3110.4103/sni.sni_295_1729527389 [Google Scholar] [CrossRef] [PubMed]
[6]. Dutta G, Singh D, Saran RK, Singh H, Srivasava AK, Jagetia A, Cerebellopontine angle anaplastic ganglioglioma masquerading as vestibular schwannoma: unusual entityWorld Neurosurg 2018 117:221-24.10.1016/j.wneu.2018.06.05429929035 [Google Scholar] [CrossRef] [PubMed]
[7]. Bram R, Seidman RJ, Chesler D, Atypical presentation of a paediatric cerebellar gangliogliomaPaediat Neurosurg 2018 53:43-48.10.1159/00047997528926844 [Google Scholar] [CrossRef] [PubMed]
[8]. Saad AF, Layton KF, Finn SS, Opatowsky MJ, Ganglioglioma and migraine headacheProc (Bayl univ Med cent) 2014 27(3):215-16.10.1080/08998280.2014.1192911324982564 [Google Scholar] [CrossRef] [PubMed]
[9]. Brainer-Lima PT, Brainer-Lima AM, Azevedo-Filho HR, Ganglioglioma comparison with other low-grade brain tumoursArq Neuropsiquiatr 2006 64(3-A):613-18.10.1590/S0004-282X200600040001817119805 [Google Scholar] [CrossRef] [PubMed]
[10]. Horbinski C, Kofler J, Yeaney G, Camelo-Piragua S, Venneti S, Louis DN, Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomasBrain Pathol 2011 21:564-74.10.1111/j.1750-3639.2011.00480.x21314850 [Google Scholar] [CrossRef] [PubMed]
[11]. Pekmezci M, Villanueva-Meyer JE, Goode B, Ziffle JV, Onodera C, Tihan T, The genetic landscape of gangliogliomaActa Neuropathologica Communications 2018 6:4710.1186/s40478-018-0551-z29880043 [Google Scholar] [CrossRef] [PubMed]
[12]. Pandita A, Balasubramaniam A, Perrin R, Shannon P, Guha A, Malignant and benign ganglioglioma: A pathological and molecular studyNeuro-Oncology 2007 9(2):124-34.10.1215/15228517-2006-02917259542 [Google Scholar] [CrossRef] [PubMed]
[13]. Kavishwar VS, Chadha KG, Barodawala SM, Murthy AK, Unusual ganglioglioma with extensive calcification and ossificationInd J Pathol Microbiol 2016 59:395-97.10.4103/0377-4929.18811527510688 [Google Scholar] [CrossRef] [PubMed]
[14]. Compton JJ, Issa Laack NN, Eckel LJ, Schomas DA, Giannini C, Meyer FB, Long-term outcomes for low-grade intracranial ganglioglioma: 30-year experience from the Mayo ClinicJ Neurosurg 2012 117:825-30.10.3171/2012.7.JNS11126022957524 [Google Scholar] [CrossRef] [PubMed]
[15]. Wolf HK, Muller MB, Spanle M, Zentner J, Schramam J, Wiestler OD, Ganglioglioma: a detailed histopathological and immunohistochemical analysis of 61 casesActa Neuropathol (Berl) 1994 88:166-73.10.1007/BF002945107985497 [Google Scholar] [CrossRef] [PubMed]
[16]. Majores M, Lehe MV, Fassunke J, Schramm J, Becker AJ, Simon M, Tumour recurrence and malignant progression of gangliogliomasCancer 2008 113:3355-63.10.1002/cncr.2396518988291 [Google Scholar] [CrossRef] [PubMed]
[17]. Bongaarts A, Prabowo A, Arena A, Anink JJ, Reinten RJ, Jansen FE, Micro RNA 519d and micro RNA 4758 can identify gangliogliomas from dysembryoplastic neuroepithelial tumours and astrocytomasOncotarget 2018 9(46):28103-15.10.18632/oncotarget.2556329963264 [Google Scholar] [CrossRef] [PubMed]
[18]. Chogule M, Pawar V, Chivate R, Ganglioglioma of CentralNervous System - study of 3 cases - with review of literatureInt J Health Sci Res 2015 5(6):720-24. [Google Scholar]