Alexander Disease (AxD) is a well-known leukodystrophy with characteristic findings on brain Magnetic Resonance Imaging (MRI) including bilaterally symmetric, frontal confluent white matter signal abnormality with relative sparing of the parietal and occipital lobes, abnormal signal in the cerebellum and brainstem, as well as swelling of the deep grey nuclei. Aside from tumour-like “masses” thought to reflect collections of Glial Fibrillary Acid Protein (GFAP), there are no additional reported associations within the central nervous system in this disease. However, in our experience authors have noted that the olfactory bulbs are more prominent in patients with AxD than in age-matched controls. Here, a 12-year-old boy with genetically confirmed AxD, with classic brain parenchymal changes and large olfactory bulbs is reported. Although the clinical implications of this finding are currently unclear, identification and reporting of this potentially associated finding may have a role in the management of patients with AxD and could be valuable for treatment of unrelated medical issues.
Brain, Leukodystrophy, Paediatrics
Case Report
A 12-year-old male presented at two years of age with developmental delays, strong right-handed dominance, and a generalised tonic-clonic seizure. MRI was performed at the time of presentation which demonstrated bilaterally symmetric, confluent T1 and T2 prolongation in the cerebral white matter with a frontal predominance, but also involving the parietal lobes and to a much lesser extent the occipital and temporal lobes. There was also T2 prolongation in the medulla, bilateral dentate nuclei, bilateral basal ganglia, and bilateral thalami. There was a periventricular rim of T1 and T2 shortening. There was no associated enhancement following contrast administration and no associated diffusion restriction. There was prominent global atrophy involving the supratentorial and infratentorial parenchyma with prominent supratentorial ventricles and sulci. There was associated thinning of the dorsal pons and midbrain with prominence of the cerebral aqueduct and fourth ventricle. All of these findings were in keeping with AxD. Diagnosis of infantile AxD was confirmed based on a genetic mutation in Exon 1 of the GFAP gene (GFAP mutation R79C).
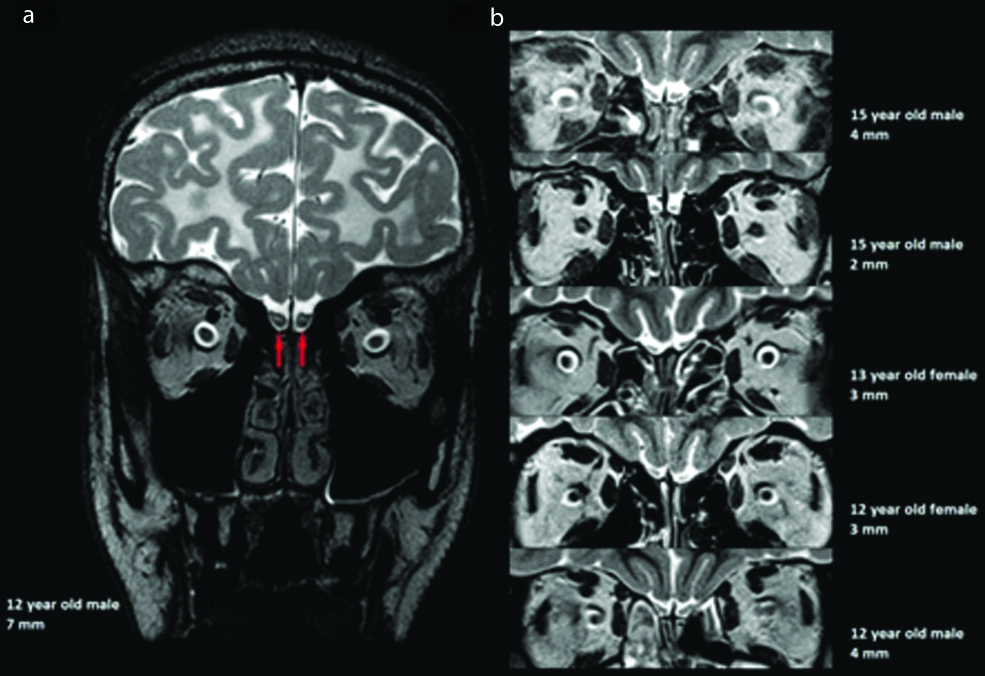
Additionally, at the time of most recent follow-up imaging, it was noted that the bilateral olfactory bulbs appeared abnormally enlarged along their entire course, without associated post-contrast enhancement. The olfactory bulbs measured 7 mm in maximum diameter on the first slice posterior to the posterior sclera [Table/Fig-1a], which was larger than any of the similarly aged-matched healthy control subjects [Table/Fig-1b]. The AxD patient demonstrated T2 prolongation in the central white matter of the bulbs. The abnormal olfactory bulbs also had a more ovoid-enlarged morphology compared to the usual J-shaped concave morphology, with resulting decreased surrounding cerebrospinal fluid.
a) Coronal T2-weighted image from the most recent follow-up MRI at the level of the olfactory bulbs on the first slice posterior to the posterior sclera in a 12-year-old male patient with Alexander disease. There is bilaterally symmetric prominence of the olfactory bulbs (red arrows), with an ovoid morphology, measuring up to 7 mm in maximum diameter. There is also T2 prolongation centrally within the olfactory bulbs bilaterally. Bilaterally symmetric confluent T2 prolongation is noted throughout the visualised white matter of the frontal lobes and the cerebral sulci are diffusely prominent in keeping with volume loss. b) Coronal T2-weighted images of the olfactory bulbs on the first slice posterior to the posterior sclera in five neurologically healthy subjects of similar age. From the top of the figure going down, the olfactory bulbs measured 4, 2, 3, 3, and 4 mm in maximum diameter in these subjects, and displayed the expected J-shaped concave morphology.
Within the five months following presentation, the patient had two additional generalised tonic-clonic seizures and was subsequently started on Keppra monotherapy. At the time of write-up of this report he was on 20 mg/kg/day and has been seizure free for almost 10 years. The only other medications he takes, are a variety of vitamins and supplements. The patients gross motor skills, fine motor skills, and language has been delayed since the time of presentation. To this day he works with a physical therapist once a week, occupational therapist once a week, and a speech therapist three days per week. He has made substantial progress in all of these areas and is currently in school, however, he remains significantly delayed globally and works one-on-one with a teaching aid.
Follow-up imaging over the course of the 10 years following diagnosis demonstrated progressive T2/FLAIR hyperintensity throughout the supratentorial white matter, over time worsening to involve all lobes completely. T2/FLAIR hyperintensity also progressively worsened in the brainstem, which by 12 years of age was nearly completely involved. Diffuse cerebral volume loss, both supra- and infratentorialy, was also noted to progressively worsen over the course of follow-up imaging.
Discussion
In this report, a potential association of abnormal olfactory bulb enlargement, with ovoid morphology and a loss of the usual J-shape, and T2 prolongation centrally with AxD, that has not been reported previously with this disorder is shown. There is a strong molecular understanding of the disease in transgenic mice that can help explain present findings.
AxD is a rare, but well known, leukodystrophy that presents most commonly as an infantile form in the first two years of life with macrocephaly, rapid neurological deterioration, seizures, spasticity, as well as behavioural, feeding, and speech problems, and delayed motor development [1,2]. Mutations in the GFAP lead to formation of Rosenthal fibres, the histologic hallmark of AxD [1-4]. The later-onset juvenile and adult forms of AxD, which exhibit longer life expectancies and motor and brainstem symptoms instead of cognitive deficits, have also been associated with GFAP mutations [2].
Van der Knaap MS et al., identified brain MR imaging criteria associated with the diagnosis of juvenile AxD [5]. These include symmetric, confluent T1-weighted and T2-weighted prolongation in the frontal white matter, T2-weighted shortening or prolongation in the basal ganglia, thalami, and brainstem, a rim of periventricular T1-weighted and T2-weighted shortening, and contrast enhancement in the periventricular rim and frontal white matter, midbrain, putamen, and head of caudate [5,6].
AxD patients accumulate ubiquitinated protein aggregates of small heat shock proteins, vimentin, and GFAP that form rods called Rosenthal fibres, which are ultimately caused by mutations in GFAP, resulting in astrocyte dysfunction [1-4]. Rather than exhibit the full spectrum of infantile AxD, GFAP mutant mice lines with the same mutations found in humans develop a less severe encephalopathy that is more analogous to the adult-onset AxD. Nevertheless, the mutant GFAP mice develop Rosenthal fibres in various brain regions and also in the olfactory bulbs [7]. Furthermore, microarray analysis of the olfactory bulbs in a different GFAP mutant mice line at three weeks and four months showed that the olfactory bulbs develop a stress response with up-regulation of genes involves iron homeostasis, glutathione metabolism, and peroxide detoxification [8]. AxD mice have also been shown to have downregulation of additional genes, Kir4.1 and GLT-1, which results in decreasing astrocytes ability to maintain homeostasis [9]. Based on this prior work of GFAP transgenic mice, it is likely that the present findings are part of the disease process and carry over to the human infantile AxD.
Understanding of olfactory bulb development also helps explains the present findings. The olfactory bulbs and nerves are not cranial nerves, but rather an extension of the telencephalic vesicles that lead to the formation of the forebrain [10,11]. The olfactory bulbs contain central white matter and an external neuronal layer. Therefore, it is not unreasonable to expect extension of the white matter pathology from the frontal lobes to the olfactory bulbs in AxD. At birth, the olfactory bulbs have a round to oval shape with central T2 prolongation from the unmyelinated white matter [10,11]. By 5.2 years of age, the olfactory bulbs develop an adult-type morphology, which is more J-shaped with more prominent lateral than central components, and demonstrates minimal central T2 prolongation [10,11]. The present authors suspect that the extension of the same ongoing supratentorial white matter abnormalities in AxD results in abnormal maturation of olfactory bulbs.
The case report recognition of enlarged abnormal olfactory bulbs in patients with infantile AxD is of uncertain clinical significance, but there are strong histologic, molecular, and MRI findings to support that this is an additional feature of the disease spectrum. Patients with AxD often have failure to thrive; the aetiology of which is multifactorial and could be attributed to dysphagia and emesis from brainstem involvement-Franzoni E et al., report a patient with juvenile AxD suffering from anorexia nervosa secondary to a pontine lesion associated with the disease [12]. It is conceivable that olfactory bulb dysfunction could alter the sense of smell, thus causing a decrease in oral intake. Although the clinical implications of olfactory bulb dysfunction are currently unclear, identification and reporting of this potentially associated finding may have a role in the management of patients with AxD but could also be important for treatment of nutritional status. Future studies should further look into this probable feature of the rare disease as it can potentially have an effect on patient quality of life.
Conclusion
Here, authors report enlarged abnormal olfactory bulbs in a patient with infantile AxD. Although of uncertain clinical significance, there is strong evidence to support this is an additional feature of the disease spectrum. Patients with AxD often have failure to thrive, which is multifactorial and may be attributable to brainstem involvement and possibly associated olfactory bulb dysfunction. Identification of this potentially associated finding may have a role in the management of patients with AxD, however, further research into this probable feature of the rare disease is necessary.
[1]. Alexander WS, Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infantBrain 1949 72(3):373-81.10.1093/brain/72.3.37315409268 [Google Scholar] [CrossRef] [PubMed]
[2]. Online Mendelian Inheritance in Man, OMIM (TM) [Internet]. Johns Hopkins University, Baltimore, MD. MIM Number: 203450; [revised 2015 Mar 12; cited 2018 Jun 10]. Available from http://www.ncbi.nlm.nih.gov/omim/ [Google Scholar]
[3]. Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A, Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander diseaseNat Genet 2001 27(1):117-20.10.1038/8367911138011 [Google Scholar] [CrossRef] [PubMed]
[4]. Green L, Berry IR, Childs AM, McCullagh H, Jose S, Warren D, Whole exon deletion in the GFAP gene is a novel molecular mechanism causing Alexander diseaseNeuropediatrics 2018 49(2):118-22.10.1055/s-0037-160892129253910 [Google Scholar] [CrossRef] [PubMed]
[5]. van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Alexander disease: diagnosis with MR imagingAJNR Am J Neuroradiol 2001 22(3):541-52. [Google Scholar]
[6]. Sarkar S, Sinha R, Chakraborty A, Khaitan T, Bhowmik B, Infantile Alexander disease: case report and review of literatureJ Clin Diagn Res 2017 11(6):ZD14-15.10.7860/JCDR/2017/26875.1010628764307 [Google Scholar] [CrossRef] [PubMed]
[7]. Hagemann TL, Connor JX, Messing A, Alexander disease glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress responseJ Neurosci 2006 26(43):11162-73.10.1523/JNEUROSCI.3260-06.200617065456 [Google Scholar] [CrossRef] [PubMed]
[8]. Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A, Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunctionHum Mol Genet 2005 14(16):2443-58.10.1093/hmg/ddi24816014634 [Google Scholar] [CrossRef] [PubMed]
[9]. Minkel HR, Anwer TZ, Arps KM, Brenner M, Olsen ML, Elevated GFAP induces astrocyte dysfunction in caudal brain regions: a potential mechanism for hindbrain involved symptoms in type II Alexander diseaseGlia 2015 63(12):2285-97.10.1002/glia.2289326190408 [Google Scholar] [CrossRef] [PubMed]
[10]. Schneider JF, Floemer F, Maturation of the olfactory bulbs: MR imaging findingsAJNR Am J Neuroradiol 2009 30(6):1149-52.10.3174/ajnr.A150119279285 [Google Scholar] [CrossRef] [PubMed]
[11]. Sarnat HB, Flores-Sarnat L, Wei XC, Olfactory development, part I: function, from fetal perception to adult wine-tastingJ Child Neurol 2017 32(6):566-78.10.1177/088307381769086728424010 [Google Scholar] [CrossRef] [PubMed]
[12]. Franzoni E, Van der Knapp MS, Errani A, Colonnelli MC, Bracceschi R, Malaspina E, Unusual diagnosis in a child suffering from juvenile Alexander disease: clinical and imaging reportJ Child Neurol 2006 21(12):1075-80.10.1177/7010.2006.0023517156703 [Google Scholar] [CrossRef] [PubMed]