Potter’s Sequence with Bilateral Renal Agenesis and Congenital Pouch Colon with Rectovaginal Fistula: A Case Report
Preeti Rai1, Reema Bhushan2, Renu Singh3, Mariya Khatoon Ansari4
1 Professor, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
2 Senior Resident, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
3 Postgraduate Student, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
4 Senior Resident, Department of Pathology, Lady Hardinge Medical College, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Reema Bhushan, 1113, Sector-4, RK Puram, New Delhi-110022, India.
E-mail: drreems25@gmail.com
Bilateral renal agenesis is an uncommon diagnosis of prenatal life. It is seen to be associated with many other congenital anomalies and is also seen in association with Potter’s sequence. A still born female baby of 38 weeks and 5 days period of gestation was delivered by Lower Segment Caesarean Section (LSCS) with physical findings of low-set ears, a flat nose, loose skin fold over the neck, wide set eyes and bilateral club foot, suggestive of Potter’s sequence. On autopsy, baby had ascitis with smaller lungs. Both the kidneys were not identified in the renal fossa or at any other (ectopic) site (bilateral renal agenesis). There was also an associated unilateral ureteral agenesis. Additional finding that was detected at autopsy was dilated terminal bowel with congenital pouch colon, rectovaginal fistula and anal atresia (anorectal malformation). This case has been reported due to rarity of the combination of these constellations of findings.
Anal atresia, Anomalies, Kidney
Case Report
A 30-year-old multigravida female was admitted in maternity ward in Lady Hardinge Medical College and Smt. Suchita Kriplani Hospital with G3P1L1A1 (38 weeks+5 days period of gestation) with breech baby, scar tenderness, bleeding per vaginum and oligohydramnios. Both the parents were Hindu. Mother was lean built.
On ultrasonography, there were multiple gross congenital anomalies in the baby. Ultrasound revealed single intrauterine foetus with reduced liquor, lung hypoplasia, and bladder like cystic structure in the abdomen along with ascites. She had a history of previous full term female baby with left eye defect and hypoplastic left ear, born eight years ago in a private local hospital and another pregnancy five years ago which resulted in miscarriage, after one and a half month amenorrhea.
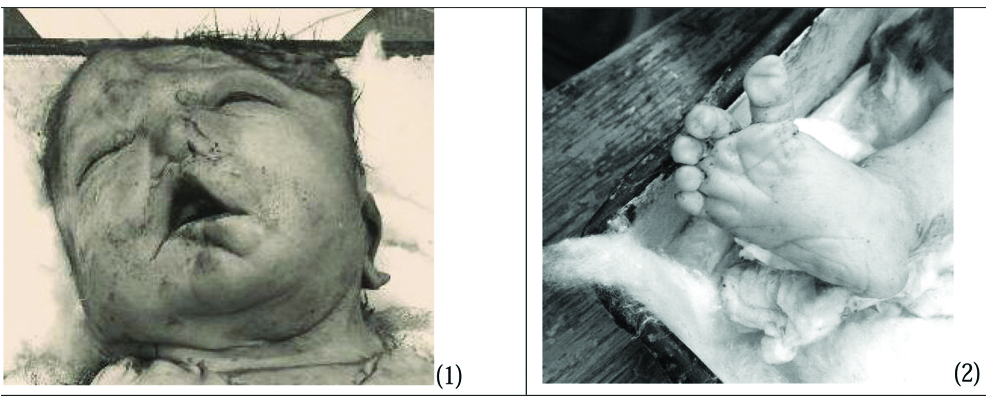
Emergency Lower Segment Caesarean Section (LSCS) was done and still born foetus was delivered. A female stillborn baby was received for autopsy in the Department of Pathology, Lady Hardinge Medical College. The weight of the baby was 2.62 kg and was 52 cm in length from head to toe. Physical examination of the stillborn baby revealed low-set ears, a flat nose, and loose skin fold over the neck, wide set eyes and bilateral club foot [Table/Fig-1,2]. External genitalia appeared normal. Umbilical cord was autolysed. All digits were present in both the feet. Anal opening, urethral and vaginal opening were externally visible. However, to check anal patency, probe was attempted to pass into the external anal opening. The probe could not be passed into the externally visible anal opening.
Physical examination of the stillborn baby revealed low-set ears, a flat nose, loose skin fold over the neck, wide set eyes and club foot.
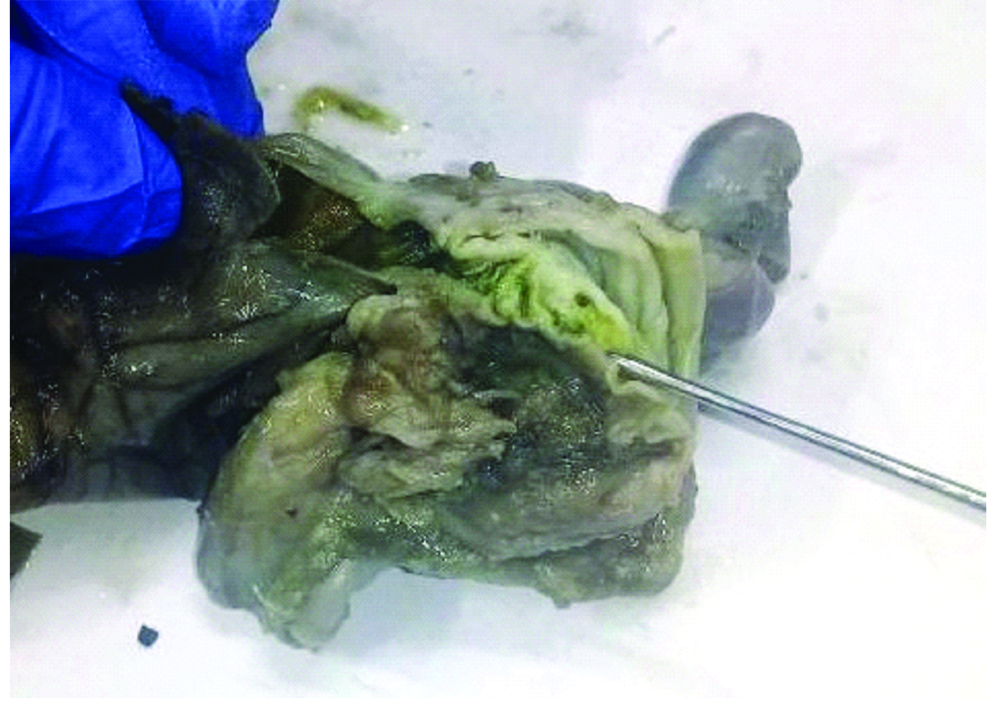
On autopsy, abdominal cavity was filled with ascitic fluid. Both lungs appeared smaller in size. Both kidneys were not identified in the renal fossa or at any other (ectopic) site (bilateral renal agenesis). However, both the adrenal glands were present. Left ureter was not identified grossly or microscopically (unilateral ureteral agenesis). Ureter was identified only on the right side which opened into the right side of the bladder. No ureteric opening was seen on the left side of the bladder. Rectum appeared patent, and dilated with thinned out wall near its terminal end and was filled with meconium (suggestive of congenital pouch colon type 4). However, the distal most rectal end appeared stenosed and grossly opened into the vagina (rectovaginal fistula; [Table/Fig-3]) which was also confirmed microscopically. The rectum was not seen connecting to the anus. Internal anal opening was not patent (anal atresia). The parents were advised to get their chromosomal analysis done to rule out the genetic factor.
Distal most bowel end appears stenosed and grossly open into vagina (rectovaginal fistula).
Discussion
Kidneys are developed as early as 12 weeks of gestation [1] located on either side of the spine in the retroperitoneal position. Absence of kidneys in renal fossa with pulmonary hypoplasia and oligohydramnios or anhydramnios strongly suggests the diagnosis of bilateral renal agenesis. It is seen in 1:10000 foetuses resulting in still birth in about 50% of the foetuses. It is generally being sporadic. However, it may have familial inheritance. It also results in death of the live born child shortly after birth. Bilateral renal agenesis is found to be caused due to lack of formation of the metanephric blastema by the ureteral bud. Newborns with bilateral renal agenesis have low-set ears, flat nose, redundant loose skin, wide set eyes, prominent fold arising at the inner canthus of each eye, parrot beak nose and receding chin. Bilateral renal agenesis is also associated with many other congenital anomalies having either autosomal dominant or recessive inheritance and is seen in association with Potter’s sequence [1,2].
‘Potter’s syndrome’ was first described by Edith Potter in newborns with bilateral renal agenesis or in association with other kidney abnormalities like renal aplasia, dysplasia, hypoplasia, or multicystic renal disease. Potter’s syndrome occurs in sporadic and autosomal recessive forms with an incidence of 1 in 4000 births [3]. A case reported by Devriendt K et al., had described a female foetus with the Potter sequence having unilateral renal agenesis, contralateral multicystic renal dysplasia, with submicroscopic deletion in chromosome 22q11 [4]. However, in the present case, parents were advised to get their chromosomal analysis done to rule out the genetic factor as there is unknown degree of recurrence risk [5], though no result was obtained till date. The male to female ratio is 2:1 [6]. However, the present case in reported in female foetus. Bilateral renal agenesis, obstruction of the urinary tract and at times rarely prolonged rupture of the membranes can lead to oligohydramnios [7]. Thus, unilateral renal agenesis is generally not seen to be associated with oligohydramnios and pulmonary hypoplasia [8]. Ureteric atresia and other lower genitourinary tract abnormalities have been described in cases reported as Potter’s syndrome [9]. Present case show agenesis of ureter on the left side. During weeks 5 to 7 of gestation there is division of the cloaca into the bladder and anorectal canal. Along with it, the development of the mullerian, wolffian and urethral structures also take place. Arrest at this stage of development may result in either or a combination of anorectal, urinary tract, genital and lower spinal anomalies [10]. Few series published showed urologic anomalies in infants with imperforate anus [11]. Urinary tract problems are generally seen as a finding in patients with imperforate anus [10].
Congenital Pouch Colon (CPC) was first reported and described by Trusler GA et al., in 1954 [12]. Cases of CPC associated with Anorectal Malformations (ARM) by a fistula communicating with the genitourinary tract [13] are reported in the literature. Many cases of CPC have been reported from India. However, few cases have also been reported in the literature from other countries like China, Japan, UK, USA, Sweden, and Saudi Arabia [14-17]. The male to female ratio have been found to be approximately 3.5:1 [15,18]. The classification of CPC was proposed by Narasimharao KL et al., which was based on the length of normal colon proximal to the colonic pouch [19]. Ileum opening directly into a pouch constitutes type 1 CPC. In type 2 CPC, ileum opens into a short segment of the cecum, which then opens into a pouch. There is at least 10 to 15 cm of normal colon between the ileum and the pouch in type 3 variant of CPC. However, in type 4 CPC, only the terminal portion of the colon (sigmoid or rectum) is converted into a pouch. The subtypes I and II are more common in both males and females [20]. Majority of the cases in the literature have reported the opening of the colonic pouch into either the vagina or in a persistent cloaca in females [14,19]. Potter’s syndrome with CPC and associated malformations is very rare in the literature. After extensive search only one case report was found in which foetus presented with Potter’s sequence, congenital pouch colon with ARM [21]. Present case showed CPC with rectovaginal fistula along with Potter’s sequence.
Conclusion
The foetus in this report, presented with Potter’s sequence having low-set ears, a flat nose, loose skin fold over the neck, wide set eyes, bilateral club foot, bilateral renal agenesis, unilateral ureteric agenesis, congenial pouch colon and rectovaginal fistula. There is no known method of prevention and the mortality rate is high. Hence, early antenatal diagnosis is important so as to allow for early, less traumatic termination of pregnancy. The evaluation of patients with the Potter’s sequence should include chromosome analysis and ultrasonographic prenatal monitoring of subsequent pregnancies.
[1]. Bronshtein M, Amit A, Achiron R, Noy I, Blumenfeld Z, The early prenatal sonographic diagnosis of renal agenesis: Techniques and possible pitfallsPrenat Diagn 1994 14(4):291-97.10.1002/pd.1970140409 [Google Scholar] [CrossRef]
[2]. Parikh H, Raniga S, Parikh N, Vaghela PP, Antenatal diagnosis of bilateral renal agenesis with potter’s sequence: A rare caseIndian Journal of Clinical Practice 2012 22(10):526-28. [Google Scholar]
[3]. Shastry SM, Kolte SS, Sanagapati PR, Potter’s SequenceJ Clin Neonatol 2012 1(3):157-59.10.4103/2249-4847.10170524027716 [Google Scholar] [CrossRef] [PubMed]
[4]. Devriendt K, Moerman P, Schoubroeck DV, Vandenberghe K, Fryns JP, Chromosome 22ql 1 deletion presenting as the Potter sequenceJ Med Genet 1997 34:423-25.10.1136/jmg.34.5.4239152843 [Google Scholar] [CrossRef] [PubMed]
[5]. Curry CJ, Jensen K, Holland J, Miller L, Hall BD, The Potter sequence: A clinical analysis of 80 casesAm J Med Genet 1984 19(4):679-702.10.1002/ajmg.13201904086393764 [Google Scholar] [CrossRef] [PubMed]
[6]. Potter EL, Facial characteristics of infants with bilateral renal agenesisAmerican Journal of Obstetrics and Gynaecology 1946 51:885-88.10.1016/S0002-9378(16)39968-9 [Google Scholar] [CrossRef]
[7]. Himabindu A, Rao BN, A fatal case of potters syndromeJournal of Clinical and Diagnostic Research 2011 5(6):1264-66. [Google Scholar]
[8]. Renal Agenesis Surveillance-United StatesMMWR 1988 37(44):679-80.:685-86. [Google Scholar]
[9]. Muren C, Wikstad I, Unilateral hydronephrosis with congenital absence of contralateral kidney in childrenActa Radiologica 1988 29(6):679-83.10.1177/0284185188029006143056472 [Google Scholar] [CrossRef] [PubMed]
[10]. Metts JC, Kotkin L, Kasper S, Genital malformations and coexistent urinary tract or spinal tract anomalies in patients with imperforate anusJ Urol 1997 158(2):1298-300.10.1016/S0022-5347(01)64460-4 [Google Scholar] [CrossRef]
[11]. Parrott TS, Urologic implications of anorectal malformationsUrologic Clinics of North America 1985 12(1):13-21. [Google Scholar]
[12]. Trusler GA, Mestel AL, Stephens CA, Colon malformation with imperforate anusSurgery 1959 45(2):328-34. [Google Scholar]
[13]. Pavai A, Pillai SD, Shanthakumari S, Sam CJ, Shylaja M, Congenital pouch colon: Increasing association with low anorectal anomaliesJ Indian Assoc Pediatr Surg 2009 14:218-20.10.4103/0971-9261.5960620419025 [Google Scholar] [CrossRef] [PubMed]
[14]. Gupta DK, Sharma S, Congenital pouch colon. In: Holschneider AM, Hutson JM, editorsAnorectal Malformations in Children: Embryology, Diagnosis, Surgical Treatment, Follow-up 2006 Berlin HeidelbergSpringer-Verlag:211-22. [Google Scholar]
[15]. Arestis NJ, Clarke C, Munra FD, Micallef C, O’Sullivan MJ, Congenital Pouch Colon (CPC) associated with anorectal agenesis: A case report and review of literaturePediatr Dev Pathol 2005 8:701-05.10.1007/s10024-005-0082-z16222474 [Google Scholar] [CrossRef] [PubMed]
[16]. Herman TE, Coplen D, Skinner M, Congenital short colon with imperforate anus (pouch colon): Report of a casePediatr Radiol 2000 30:243-46.10.1007/s00247005073010789902 [Google Scholar] [CrossRef] [PubMed]
[17]. Donkol RH, Jetley NK, Al Mazkary MH, Congenital pouch colon syndrome in a Saudi Arabian neonateJ Pediatr Surg 2008 43:e9-e11.10.1016/j.jpedsurg.2007.08.06618206446 [Google Scholar] [CrossRef] [PubMed]
[18]. Shakya VC, Agrawal CS, Koirala R, Khaniya S, Poudel P, Adhikary SS, A report of a rare congenital malformation in a Nepalese child with congenital pouch colon: a case reportCases J 2009 2:6424doi: 10.1186/1757-1626-0002-000000642410.1186/1757-1627-2-6447-2-6424 [Google Scholar] [CrossRef]
[19]. Narasimharao KL, Yadav K, Mitra SK, Congenital short colon with imperforate anus (pouch colon syndrome)Ann Pediatr Surg 1984 1:159-67. [Google Scholar]
[20]. Chadha R, Khan NA, Congenital pouch colonJ Indian Assoc Pediatr Surg 2017 22:69-78.10.4103/jiaps.JIAPS_5_1728413299 [Google Scholar] [CrossRef] [PubMed]
[21]. Supriya G, Saritha S, Suseelamma D, Kumar PM, Potter’s syndrome associated with pouch colon anomaly in exomphalosAnat Physiol 2012 2:110doi:10.4172/2161-0940.100011010.4172/2161-0940.1000110 [Google Scholar] [CrossRef]