The Constitutional Balanced Translocation t(11;22)(q23;q11.2)-An Indian Account
Vandana Kamath1, Vivi M Srivastava2, S Yuvarani3, Mary Purna Chacko4, Saurabh Kumar Bhattacharya5, Samuel Phillip Oommen6, Danda Sumita7, George Korula8
1 Associate Professor, Department of Cytogenetics, Christian Medical College, Vellore, Tamil Nadu, India.
2 Professor and Head, Department of Cytogenetics, Christian Medical College, Vellore, Tamil Nadu, India.
3 Technologist, Department of Cytogenetics, Christian Medical College, Vellore, Tamil Nadu, India.
4 Professor, Department of Cytogenetics, Christian Medical College, Vellore, Tamil Nadu, India.
5 Tutor, Department of Cytogenetics, Christian Medical College, Vellore, Tamil Nadu, India.
6 Professor, Department of Developmental Paediatrics, Christian Medical College, Vellore, Tamil Nadu, India.
7 Professor, Department of Medical Genetics, Christian Medical College, Vellore, Tamil Nadu, India.
8 Professor, Department of Reproductive Medicine Unit, Christian Medical College, Vellore, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Vivi M Srivastava, Professor and Head, Christian Medical College, Ida Scudder Road, Vellore-632004, Tamil Nadu, India.
E-mail: cytogen@cmcvellore.ac.in
Introduction
The balanced translocation t(11;22) is one of the most common constitutional genetic abnormality detected in humans. Carriers of the t(11;22) are usually phenotypically normal and their carrier status is ascertained only if they present with reproductive losses, infertility or a child with an abnormal phenotype. The t(11;22) translocations are a cumulative effect of recombination errors occurring during meiosis. Children with Emanuel syndrome show a gain of additional genetic material in the form of der(22) chromosome on conventional karyotype inherited either from the carrier parents or de novo in origin. Children with der(22) chromosome presented with microcephaly, hypotonia, preauricular sinus and developmental delay.
Aim
To study the mode of inheritance and outcome of the balanced translocation t(11;22) in the families.
Materials and Methods
A total of 16 individuals from six unrelated families underwent cytogenetic analysis at the Christian Medical College, Vellore, Tamilnadu, India, and their karyotype showed the balanced t(11;22)(q23;q11.2) or a der(22)t(11;22)(q23;q11.2) inherited from a t(11;22) carrier parent. Also, karyogram of the spouses of the carrier individuals were also studied. Conventional cytogenetic analysis of phytohaemagglutinin-stimulated peripheral blood cultures was performed. Fluorescence In Situ Hybridization (FISH) was performed to confirm the der(22) t(11;22)(q23;q11.2).
Results
Seven individuals from six unrelated families showed the balanced t(11;22). These included six adults and one child (five females and two males). All six adult carriers were phenotypicaly normal. In three adults, the translocation was ascertained because their children had abnormal phenotypes. The remaining three adults were from families being investigated for recurrent pregnancy losses. One of these subsequently underwent amniocentesis which showed a translocation morphologically identical to that in the father. All the three carrier parents had children with abnormal phenotypes. And their karyogram showed 47 chromosomes due to gain of a supernumerary chromosome+der(22)t(11;22)(q23;q11.2) of maternal origin, while the fourth child presented with a t(11;22) identical to that of her carrier father, but was lost to follow-up.
Conclusion
It is important to be aware of this balanced translocation and its varied outcomes, so that members of the family can be studied. This would help to determine the mode of inheritance and to predict the likelihood of other carriers in the family having children with chromosomal imbalance.
Emanuel syndrome, Meiosis, Recombination, Recurrent pregnancy losses, Segregation
Introduction
Constitutional balanced translocations are seen in about 1 in 600 live births [1]. Some of these translocations are de novo, while others are inherited from a parental balanced translocation and may be seen in more than one member of a family. These translocations are a result of errors in recombination during meiosis [2,3].
Carriers of balanced translocations are usually normal and are identified only if they have reproductive losses, infertility or a child with an abnormal phenotype. These adverse effects occur if the manner of segregation of chromosomes into gametes results in partial trisomy for one of the chromosomes involved in the translocation and partial monosomy for the other (segmental aneusomy). A conceptus with segmental aneusomy will proceed to term only if the segments involved are small; if the segments involved in the translocation are large, the resulting genetic imbalance would be lethal to the conceptus [4]. The t(11;22)(q23;q11.2) carrier parents may have offspring with an abnormal phenotypes due to a gain of der(22)t(11;22)(q23;q11.2) chromosome, which is known as supernumerary der(22)t(11;22)q23;q11.2) syndrome or more commonly termed as Emanuel syndrome. Gain of a supernumerary der(22)t(11;22) may be the result of a de novo occurrence in a small subset of children. Emanuel syndrome is a unique syndrome which is characterised by constellation of multiple congenital malformations, significant developmental delay which is evident in almost 100% of children and distinct craniofacial dysmorphism. The abnormalities of pinna in the form of a perauricular sinus/tag are pathognomonic of Emanuel syndrome [5].
Less commonly, the translocation may disrupt a gene or its regulatory elements, or cause a position effect in which the relocation of a gene as a result of the translocation interferes with its functioning [6,7].
Most of these translocations are unique to the members of a particular family. Only three recurrent translocations have been reported till date, namely, the t(11;22) (q23.3;q11.2), the t(4;8)(p16;p23) and more recently, a t(8;22)(q24.1;q11.21) [7]. Among these, the balanced t(11;22) is the most common and has been reported in nearly 200 individuals from multiple unrelated families in several countries between 1980 and 1983 [8,9].
In this study, we karyotyped 16 individuals from six unrelated families, of which seven presented with the t(11;22)(q23;q11.2), three with der(22)t(11;22)(q23;q11.2) which originated from carrier parents, while the remaining six were normal spouse, who were a part of the family work up, to illustrate the outcomes of the t(11;22)(q23;q11.2) and discuss its origin.
Materials and Methods
The study group consisted of 16 subjects which included six couples and four children (with three affected children and one additional child from the carrier couple) from six unrelated families, all of these individuals underwent cytogenetic analysis at the Christian Medical College, Vellore between 2001 and 2018. Seven of them showed the t(11;22)(q23;q11.2) upon karyotypic analysis (six spouse and one child), while three showed der(22)t(11;22)(q23;q11.2) chromosome inherited from a balanced t(11;22) carrier parent and the remaining six spouses of these individuals showed a normal karyotype who were a part of the family work up. The clinical presentations were retrieved from medical records. Conventional cytogenetic analysis of phytohaemagglutinin-stimulated peripheral blood cultures was performed using standard protocols [10]. Karyotypes were reviewed and recorded in accordance with the International System for Human Cytogenomic Nomenclature [11]. This retrospective study was performed in accordance with the ethical standards of the responsible committee on human experimentation. For each patient at least 20 G-banded metaphases were studied using a Zeiss Axioskop 40 microscope and an automated karyotyping system (Ikaros, Metasystems GumBy, Altlussheim, Germany). Flourescence in situ hybridization was performed in all cases for confirmation of the der(22)t(11;22)(q23;q11.2) chromosome.
Results
There were seven individuals with the balanced t(11;22) [Table/Fig-1]. These included five females and two males. Six were adults with normal phenotypes. In three adults (Unique Identification Number (UID) 1-3), the translocation [Table/Fig-2a] was ascertained because their children had abnormal phenotypes with 47 chromosomes due to a small supernumerary chromosome [Table/Fig-2b]. The remaining three adults (two men and one woman, UID 4-6) were from families being investigated for recurrent (two to six) pregnancy losses. One of these couples subsequently conceived and amniocentesis showed a translocation morphologically identical to that seen in the carrier father (UID 4); this couple, who had six pregnancy losses, elected to continue the pregnancy.
The inheritance pattern of the t(11;22)(q23;q11.2).
| Carrier | Sex | Age | Karyotype of carrier | Mode of ascertainment of translocation |
|---|
| UID 1 | F | 25 | 46,XX,t(11;22)(q23;q11.2) | Female child with an abnormal phenotype: 47,XX,+der(22)t(11;22)(q23;q11.2)mat |
| UID 2 | F | 26 | 46,XX,t(11;22)(q23;q11.2) | Male child with an abnormal phenotype: 47,XY,+der(22)t(11;22)(q23;q11.2)mat |
| UID 3 | F | 33 | 46,XX,t(11;22)(q23;q11.2) | Female child with an abnormal phenotype: 47,XX,+der(22)t(11;22)(q23;q11.2)mat |
| UID 4 | M | 26 | 46,XY,t(11;22)(q23;q11.2) | Six pregnancy losses. Amniocentesis of a subsequent pregnancy showed the t(11;22)(q23;q11.2).Female child (phenotype could not be established): post-natal karyotype-46,XX,t(11;22)(q23;q11.2)pat |
| UID 5 | M | 34 | 46,XY,t(11;22)(q23;q11.2) | Two pregnancy losses. No living children. |
| UID 6 | F | 20 | 46,XX,t(11;22)(q23;q11.2) | Three pregnancy losses. No living children. |
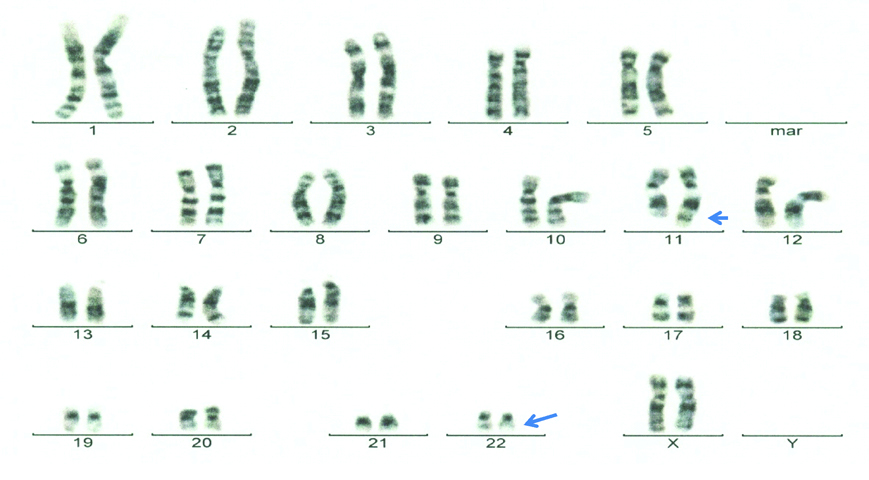
G banded karyogram (UID: 2) depicting the balanced translocation t(11;22)(q23;q11.2).
Arrow indicates the site of translocation t(11;22)(q23;q11.2) on the chromosome 11 and 22 in carrier mother.
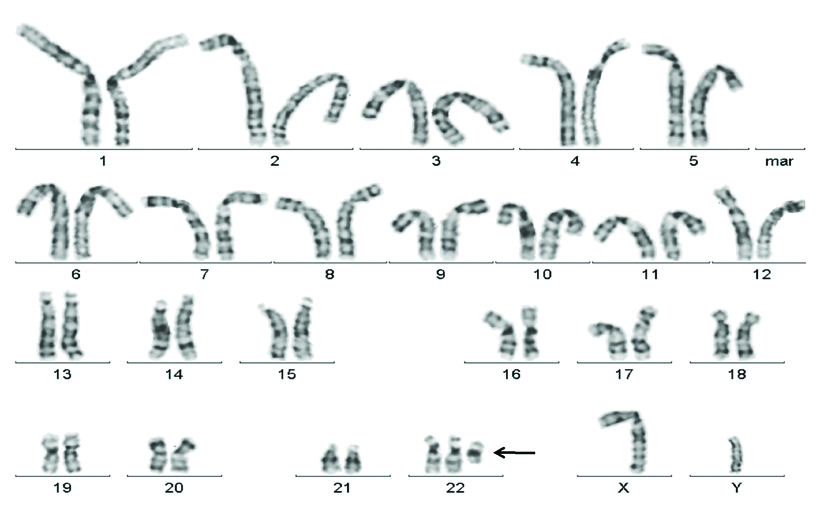
G banded karyogram (UID:2) showing the der(22)t(11;22)(q23;q11.2) chromosome.
Arrow indicates the additional der(22)t(11;22)(q23;q11.2) chromosome in the male child.
All three carrier parents (UID 1-3) had children with dysmorphism, developmental delay and congenital abnormalities, namely, cardiac septal defects (two atrial and one ventricular), hearing impairment, seizures (two each), hypotonia, visual loss and genital abnormalities [Table/Fig-3]. The karyotypes of all three showed 47 chromosomes due to gain of an abnormal (derivative) chromosome 22 (Emanuel syndrome, supernumerary chromosome 22) inherited from a maternal translocation (11;22) [Table/Fig-4]. The postnatal karyotype of the fourth child, whose translocation was detected prenatally, showed 46 chromosomes with an apparently balanced t(11;22) similar to that of her father and no additional cytogenetic abnormalities. This child however was lost to follow-up. Members of the extended families including the siblings and parents of the adult carriers were not available for study.
Clinical presentation of children with abnormal phenotype.
| Case | Age | Sex | Karyotype | Clinical Features |
|---|
| Child of UID-1 | 6 years | F | 47,XX,+der(22)t(11;22)(q23;q11.2)mat | Central hypotonia, seizures, developmental delay, dysmorphism, cleft palate, ventricular septal defect with mild coarctation, auditory and visual insufficiency, malrotation of gut |
| Child of UID-2 | 8 months | M | 47,XY,+der(22)t(11;22)(q23;q11.2)mat | Developmental delay, Dandy-Walker malformation, atrial septal defect, abnormal testis, microtia, hearing impairment |
| Child of UID-3 | 3 years | F | 47,XX,+der(22)t(11;22)(q23;q11.2)mat | Developmental delay, microcephaly, seizures, hypertelorism, atrial septal defect |
| Child of UID-4 | Newborn | F | 46,XX,t(11;22)(q23;q11.2)pat | Prematurity |
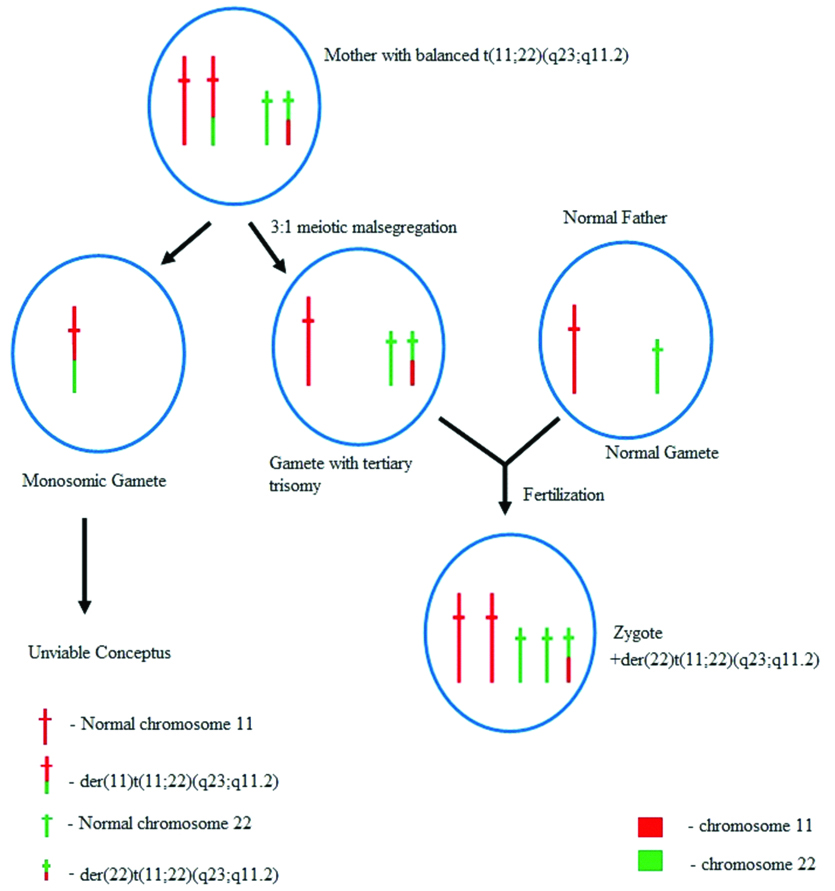
The t(11;22) is prone to 3:1 meiotic segregation during gametogenesis.
The 3:1 meiotic malsegregation occurs in meiosis I of gametogenesis with one gamete receiving three homologues (tertiary trisomy) {normal chromosomes 11 and 22 and a der(22)t(11;22)}. While the other gamete receives only one abnormal chromosome 11 (not a viable gamete). The gamete with the tertiary trisomy, if fertilizes with a normal gamete (meiosis II) results in the formation of a zygote with two normal chromosomes 11 and 22, along with a gain of a der(22) chromosome resulting in Emanuel syndrome.
Discussion
The t(11;22) is the most common recurrent non-Robertsonian constitutional translocation in humans, which has been reported in nearly 200 families from different countries till date [8,9]. These families are unrelated and do not appear to have a common ancestor.
The recurrence of the translocation depends on the genomic architecture of the regions of the breakpoints. Both chromosomes 11 and 22 have Low Copy Repeat (LCR) sequences characterised by several hundred base pairs of palindromic AT-rich repeats [12,13]. These Palindromic A-T Rich Regions (PATRRs) are a potential source of genomic instability since they have a tendency to form hairpin-like or cruciform secondary structures, the tips of which are prone to breakage [13,14]. The work of Kurahashi H et al., suggest that cruciform structures may be formed when the PATRRs in single-stranded DNA show intra-strand base pairing [15]. The PATRRs on chromosomes 11 and 22 show variations in length which further determine the likelihood of their breakage. Longer PATRRs appear to be more prone to form cruciform structures and undergo breakage than those involving shorter sequences [16]. Another factor that could be predisposed to breakage is asynchrony of DNA replication; the LCRs on chromosome 22 show both early and late replicating regions with the transition zone between these regions being prone to double-stranded breaks [17]. These double-stranded breaks in DNA predispose the regions to deletions and/or duplications as well as translocations with other chromosomes that have similar PATRRs [18]. Such translocations could be formed due to non-homologous end-joining of the double-strand breaks [14]. Ashley T et al., documented that the spatial proximity between chromosomes 11 and 22 also make them predisposed to recombination between these two chromosomes [2].
Most de novo t(11;22) are of paternal origin. This translocation has been detected at a high frequency (1/104-1/105) in sperms from normal healthy males suggesting that the generation of the t(11;22) is linked to gametogenesis [13,19]. This translocation is not found in somatic cells or cell lines [14,19].
The majority of translocation carriers has normal phenotypes and is unaware that they have a translocation until they seek medical attention because of recurrent abortions, infertility or after the birth of a child with chromosomal imbalance. During meiosis, one member of each chromosomal homologue segregates randomly into different gametes (2:2 segregation). If both the derivative (translocated) chromosomes segregate into the same gamete, a zygote formed from such a gamete will carry the balanced translocation. However, if, one normal homologue and one derivative chromosome segregate into the same gamete, the resulting zygote will have chromosomal imbalance with monosomy for one of the segments involved and trisomy for the other. The viability of zygotes with chromosomal imbalance will depend on the degree of segmental aneusomy. The degree of autosomal imbalance that can support a viable pregnancy is a loss of upto 2% of the genome for autosomal monosomies and gain of upto 4% of the genome for autosomal trisomies [4]. Therefore, carriers of such translocations are at risk of having live born infants with chromosomal imbalance. If however, the translocation involves large segments of one or both chromosomes, the degree of genetic imbalance will render the conceptus unviable, leading to reproductive loss. Carriers of such translocations will therefore be at risk of recurrent abortions or infertility and any live-born children will be phenotypically normal. A 3:1 malsegregation may also occur in meiosis I with one gamete receiving three homologues, and the other receiving only one. This could result in the zygote having an extra structurally abnormal (supernumerary) chromosome and a live-born child with chromosome imbalance. This outcome may be seen with the t(11;22) because the small size of chromosome 22 predisposes to 3:1 segregation. The resulting supernumerary chromosome would have sequences from both chromosomes 11 and 22 in addition to one normal homologue each of chromosomes 11 and 22 resulting in partial trisomies for these two chromosomes (tertiary trisomy). Ashley T et al., showed that the supernumerary chromosome 22 associated with the t(11;22) was indeed due to 3:1 malsegregation in meiosis I [2]. This form of malsegregation which results in a supernumerary chromosome is compatible with live birth only because of the relatively smaller segments being involved in the t(11;22).
The newborn who had an apparently balanced translocation similar to that of her father was lost to follow-up. Therefore, the phenotype of this child could not be ascertained. Individuals with an apparently balanced translocation can present with an abnormal phenotype either due to a submicroscopic deletion and/or duplication, a position effect, mosaicism for balanced/unbalanced translocation or a factor unrelated to the balanced translocation. DNA microarray analysis becomes necessary to determine the nature and degree of chromosomal imbalance in such scenarios.
The group of adult carriers under study, who are phenotypically normal illustrates the adverse effects of the t(11;22) which are due to the manner of segregation during meiosis, namely, recurrent abortions/infertility or having offspring with abnormal phenotypes due to chromosomal imbalance. An adult carrier can have offspring with an apparently balanced translocation, morphologically similar to the parent but with an abnormal phenotype: although the likelihood of such an outcome is low, this must be borne in mind when counselling carriers about the risk of having children with abnormal phenotypes. An another report also relates the development of the familial breast cancer to families with this translocation; however, more studies are needed to confirm this association [20].
Carriers who are ascertained through the birth of a child with chromosomal imbalance have a 5-10% risk of recurrence in a subsequent pregnancy [6,9]. In female carriers of the t(11;22), the risk of having children with the supernumerary derivative chromosome 22 syndrome is nearly 4%, while the risk figure for male carriers is 0.7% [20]. Awareness of this translocation and its mode of inheritance will help to prevent the devastating consequences of chromosomal imbalance in affected families [21].
Conclusion
It is important to be aware of this translocation, its inheritance pattern and the diverse effects, so that parents and/or siblings and members of the extended family can be studied. This would help to predict the likelihood of other carriers in the family having children with chromosomal imbalance as a result of this translocation.
[1]. Ou Z, Stankiewicz P, Xia Z, Breman AM, Dawson B, Wiszniewska J, Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomesGenome Res 2011 21:33-46.10.1101/gr.111609.11021205869 [Google Scholar] [CrossRef] [PubMed]
[2]. Ashley T, Gaeth AP, Inagaki H, Seftel A, Cohen MM, Anderson LK, Meiotic recombination and spatial proximity in the etiology of the recurrent t(11;22)Am J Hum Genet 2006 79(3):524-38.10.1086/50765216909390 [Google Scholar] [CrossRef] [PubMed]
[3]. Shaffer LG, Lupski JR, Molecular mechanisms for constitutional chromosomal rearrangements in humansAnnu Rev Genet 2000 34:297-329.10.1146/annurev.genet.34.1.29711092830 [Google Scholar] [CrossRef] [PubMed]
[4]. Firth HV, Hurst JA, Oxford Desk Reference: Clinical Genetics and Genomics (Oxford Desk Reference Series) 2017 2nd edOxford University press10.1093/med/9780199557509.001.0001 [Google Scholar] [CrossRef]
[5]. Carter MT, St Pierre SA, Zackai EH, Emanuel BS, Boycott KM, Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individualsAm J Med Genet A 2009 149A(8):1712-21.10.1002/ajmg.a.3295719606488 [Google Scholar] [CrossRef] [PubMed]
[6]. Kleinjan DJ, van Heyningen V, Position effect in human genetic diseaseHum Mol Genet 1998 7(10):1611-18.10.1093/hmg/7.10.16119735382 [Google Scholar] [CrossRef] [PubMed]
[7]. Gersen SL, Keagle MB, The Principles of Clinical Cytogenetics 2013 3rd edSpringer:143:16210.1007/978-1-4419-1688-4 [Google Scholar] [CrossRef]
[8]. Hill AS, Foot NJ, Chaplin TL, Young BD, The most frequent constitutional translocation in humans, the t(11;22)(q23;q11) is due to a highly specific alu-mediated recombinationHum Mol Genet 2000 9(10):1525-32.10888603 [Google Scholar] [CrossRef] [PubMed]
[9]. Kee SK, See VH, Chia P, Tan WC, Tien SL, Lim ST, Differential outcomes in an extended family with constitutional t(11;22)(q23.3;q11.2)J Pediatr Genet 2013 2(1):37-41. [Google Scholar]
[10]. Arsham MS, Barch MJ, Lawce HJ, The AGT Cytogenetics Laboratory Manual 2016 4th edNew JerseyJohn Wiley & Sons:98-108.:280-81. [Google Scholar]
[11]. McGowan-Jordan J, Simon A, Schmid M, An International System for Human Cytogenomic Nomenclature (2016) 2016 S. Karger, Basel [Google Scholar]
[12]. Kato T, Kurahashi H, Emanuel BS, Chromosomal translocations and palindromic AT-rich repeatsCurr Opin Genet Dev 2012 22(3):221-28.10.1016/j.gde.2012.02.00422402448 [Google Scholar] [CrossRef] [PubMed]
[13]. Kurahashi H, Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangementsClinical Genetics 2010 78(4):299-309.10.1111/j.1399-0004.2010.01445.x20507342 [Google Scholar] [CrossRef] [PubMed]
[14]. Inagaki H, Ohye T, Kogo H, Kato T, Bolor H, Taniguchi M, Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humansGenome Res 2009 19(2):191-98.10.1101/gr.079244.10818997000 [Google Scholar] [CrossRef] [PubMed]
[15]. Kurahashi H, Inagaki H, Yamada K, Ohye T, Taniguchi M, Emanuel BS, Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocationsThe Journal of Biol Chem 2004 279(34):35377-83.10.1074/jbc.M40035420015208332 [Google Scholar] [CrossRef] [PubMed]
[16]. Tong M, Kato T, Yamada K, Inagaki H, Kogo H, Ohye T, Polymorphisms of the 22q11.2 breakpoint region influence the frequency of de novo constitutional t(11;22)s in spermHum Mol Genet 2010 19(13):2630-37.10.1093/hmg/ddq15020392709 [Google Scholar] [CrossRef] [PubMed]
[17]. Puliti A, Rizzato C, Conti V, Bedini A, Gimeli G, Barale R, Low-copy repeats on chromosome 22q11.2 show replication timing switches, DNA flexibility peaks and stree inducible asynchrony, sharing instability features with fragile sitesMutat Res 2010 686(1-2):74-83.10.1016/j.mrfmmm.2010.01.02120138061 [Google Scholar] [CrossRef] [PubMed]
[18]. Kato T, Inagaki H, Tong M, Kogo H, Ohye T, Yamada K, DNA secondary structure is influenced by genetic variation and alters susceptibility to de novo translocationMol Cytogenet 2011 4:1810.1186/1755-8166-4-1821899780 [Google Scholar] [CrossRef] [PubMed]
[19]. Ohye T, Inagaki H, Kogo H, Tsutsumi M, Kato T, Tong M, Paternal origin of the de novo constitutional t(11;22)(q23;q11)Eur J Hum Genet 2010 18(7):783-87.10.1038/ejhg.2010.2020179746 [Google Scholar] [CrossRef] [PubMed]
[20]. Wieland I, Muschke P, Volleth M, Röpke A, Pelz AF, Stumm M, High incidence of familial breast cancer segregates with constitutional t(11;22)(q23;q11)Genes Chromosomes Cancer 2006 45(10):945-49.10.1002/gcc.2035816845657 [Google Scholar] [CrossRef] [PubMed]
[21]. Gardner RJM, Sutherland GR, Shaffer LG, Chromosome Abnormalities and Genetic Counseling 2012 4th edNew YorkOxford:102-107.10.1093/med/9780195375336.001.0001 [Google Scholar] [CrossRef]