Materials and Methods
The study was conducted in a 2400 bedded tertiary care hospital in southern India, over a six month period from May 2017 to October 2017. A prospective study design was adopted. Clearance was obtained from the Institutional Review Board (IRB) and Ethics Committee (Ref: IRB No 10915). S. maltophilia isolates from blood and ETA samples (with counts >105 CFU/mL) causing nosocomial infections in patients admitted to the in-patient wards where treatment was initiated for the same were included in the study. All other isolates of S. maltophilia which did not satisfy the above criteria were excluded. A total of 26 isolates were obtained during the study period and 18 of them satisfied the inclusion criteria; eight of them from blood cultures and 10 from ETA.
Identification and AST in the Laboratory
S. maltophilia is a non-fermenting Gram negative bacterium which is oxidase negative. Preliminary screening media consisting of mannitol motility medium, triple sugar iron agar, peptone water and citrate show no change in the medium except for motility being positive. Identification of such colonies which are non-lactose fermenting on MacConkey medium was carried out using Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) (bioMerieux). Antimicrobial Susceptibility Testing (AST) was performed using disc diffusion as per the recommendations in CLSI guidelines, M100-S27 (CLSI 2017).
DNA extraction: Overnight bacterial cultures grown on blood agar culture plates were emulsified in the sterile saline solution and the genomic DNA isolation was carried out by QiaSymphony (Automated DNA Extraction), as per manufacturer’s instructions.
Multi Locus Sequence Typing (MLST)
Conventional MLST (cMLST) was performed using primers and thermal profile conditions for amplification as specified in the PubMLST database (https://pubmlst.org/) using 3500 genetic analyser (Applied Biosystems, CA, USA). Allele sequence identified through cMLST were BLAST matched with NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Whole Genome Sequencing (WGS) and Analysis
Whole-Genome Sequencing (WGS) was performed using IonTorrent PGM (Life Technologies, CA, USA) with 400 bp chemistry for a subset of nine S. maltophilia strains (B27164, B23119, B27671, B26854, B09516, S04330, B26847, S04501 and B27675). Sequence types of these tested isolates were identified using allele sequences identified by WGS (wgMLST) using MLST 1.8 tool (https://cge.cbs.dtu.dk/services/MLST/). WGS based SNP phylogeny was calculated using CSI Phylogeny 1.4 tool (https://cge.cbs.dtu.dk/services/CSIPhylogeny/). This Whole Genome Shotgun project was deposited at GenBank under the accession numbers PXIJ00000000, PXIO00000000, PXIL00000000, PXIN00000000, PXIK00000000, PXII00000000, PXIM00000000, PXJF00000000 and PXJG00000000. The version described in this manuscript is version 1.
Results
Patient and Isolate Details
The patients’ age group ranged from nine months to 84 years. Pre-existing comorbid conditions in terms of chronic immunocompromised state was seen in all of them and the same is represented in [Table/Fig-1]. Only a single isolate has been taken into consideration for study purpose in cases where S. maltophilia was recovered from multiple specimens of the same patient. All blood specimens with S. maltophilia were mono-microbial; seven of the ETA had other organisms growing along with S. maltophilia in significant numbers (four Pseudomonas spp, two Acinetobacter spp and one S. aureus). All were suspected to have pathogenic potential based on X rays, blood counts, CRP, and clinical presentation and treatment was initiated.
Pre-existing comorbid conditions seen in the study patients.
| Comorbidity | N | % |
|---|
| Malignancies | 5 | 27 |
| Chronic renal failure | 7 | 38 |
| Chronic liver disease | 2 | 11 |
| *Others | 4 | 22 |
*included congenital heart disease, diabetes, hypothyroidism.
Antimicrobial Susceptibility
As expected all isolates were resistant to carbapenems. The antimicrobial susceptibility profile of the isolates is shown in [Table/Fig-2]. Clinical records showed that 12 (66%) of them received intravenous levofloxacin and the others, trimethoprim-sulfamethoxazole. Fourteen patients showed significant improvement. Two patients were discharged at request due to worsening clinical condition and two succumbed to the infection.
Antibiotic susceptibility pattern observed in the isolates of S. maltophilia (n=18).
| Drug | Resistant (n) | Intermediate (n) | Susceptible (n) |
|---|
| TMT-SMT | 1 | - | 17 |
| Levofloxacin | 0 | - | 18 |
| Ceftazidime | 12 | - | 6 |
| Tetracycline | 10 | 1 | 7 |
MLST
To identify the clonal relatedness of these isolates, conventional MLST (cMLST) was performed. For 17 out of 18 isolates, MLST PCR and sequencing was not successful for either one or more genes out of seven house-keeping genes even after several attempts. One isolate, S04384 was identified as ST13 by cMLST.
Whole Genome Sequencing
WGS was performed on a subset of nine isolates from the remaining 17 isolates on which cMLST was not successful. Analysis of WGS results using K-merFinder 2.5 tool (https://cge.cbs.dtu.dk/services/KmerFinder/) confirmed that all nine sequenced isolates were S. maltophilia. Sequences were analysed through MLST Finder and revealed the sequence type for one isolate (B27164) to be ST15. Other eight isolates were observed as novel sequence types. Most of the alleles in these eight WGS isolates did not match accurately with the allele sequences from the database (98-99% identity). The alleles with varied sequences were submitted to the PubMLST database to obtain new allele types and sequence types. As a result, four novel sequence types (ST283, ST284, ST285 and ST286) of S. maltophilia were identified using whole genome MLST (wgMLST). However, whole-genome SNP phylogeny revealed bifurcating and multifurcating groups among the nine S. maltophilia isolates. None of the isolates belonged to the same clonal group as revealed by SNP based phylogeny.
Discussion
Much of the literature on S. maltophilia available from India is in the form of case reports. To our knowledge this is the first study employing molecular typing methods on S. maltophilia from India. Worldwide many genotypic molecular typing approaches and targets have been evaluated for their discriminatory power to identify the clonal relatedness of S. maltophilia; these included ribotyping, DNA-DNA hybridizations, Amplified Fragment Length Polymorphism (AFLP), Random Amplified Polymorphic DNA (RAPD), Restriction Fragment Length Polymorphisms (RFLP) of the gyrB gene, Pulsed-Field Gel Electrophoresis (PFGE), and Enterobacterial Repetitive Intergenic Consensus Sequence-PCR (ERIC-PCR) [2]. The details of the findings on evaluation of different methodologies on discriminatory ability and robustness of aforementioned methods are given in [Table/Fig-3] [5-16].
Various typing methods with discriminatory ability for clonal relatedness of S. maltophilia [5-16].
| Typing method | Discriminatory ability of the methods | Remarks | References |
|---|
| Conventional typing | AFLP | Diversity between environmental and human clinical samples of S. maltophilia was observed with variation of 60% | Reproducibility is lowReference available is not standard and lacks global comparison | Hauben L et al., [7] |
| RAPD and ERIC-PCR | First use in S. maltophilia observed high diversity (76%) among infected patients | Chatelut M et al., [8]; Caylan R et al., [9]. |
| RFLP | 191 isolates revealed 9 clusters with internal similarity >75%Demonstrated the antimicrobial resistance due to changes in S. maltophilia strains over a period of 15 months in same patient | Wust J et al., [10]; Coenye T et al., [11] |
| PFGE | 139 strains formed 5 clusters and 71% of isolates showed dissimilar banding patternHigh diversity of strain relatedness in hospital sources is noted | Helpful in defining an outbreak from smaller population i.e., hospital outbreaksMost discriminatory among the conventional methods | Berg Get al., [12]; Valdezate S et al., [13]; Jumaa PA et al., [14] |
| Sequence based methods | MLST | 54 STs observed from 70 strains (77%)Recombination events were detected in one-sixth of the sequence types | Resulted in diverse new sequence/allele types in every analysis | Kaiser S et al., [5]; Cho HH et al., [6] |
| WGS | Identified significant genome diversity in the isolates | Accurate identification of allele types using complete sequences | Pak TR et al., [15] |
| SNP based phylogeny | Lineages were reported based on 334 polymorphic mutationsSNP analysis proved that mutations arising independently in two lineages co-localise in space, providing evidence for location-specific selection in human body | Would help in analysing the closeness of the clinical isolates and in identifying the frequency of recombination events | Chung H et al., [16] |
It is evident that surveillance of S. maltophilia using MLST platform often assigns novel ST and allele types which makes it difficult to define the clonal groups [5,6]. This is due to high frequency of recombination events naturally seen in S. maltophilia [17]. Interestingly, in this study the allele numbers for few genes assigned by cMLST (uses partial target region) were not correlating with wgMLST (complete allele sequence). Cho HH et al., had reported that out of 33 clinical isolates, 28 isolates exhibited 23 new sequence types (STs), whereas only five isolates exhibited previously described STs (3 STs) [6].
Way forward to the existing limitation is the whole genome based SNP analysis. This might give much better insights into the phylogenies of S. maltophilia. Lira F et al., had reported that the complex diversity of S. maltophilia formed an interlaced taxon and shared the same attributes between clinical and environmental strains [18]. In this study, SNP based phylogeny revealed that every study isolates is divergent from the other [Table/Fig-4].
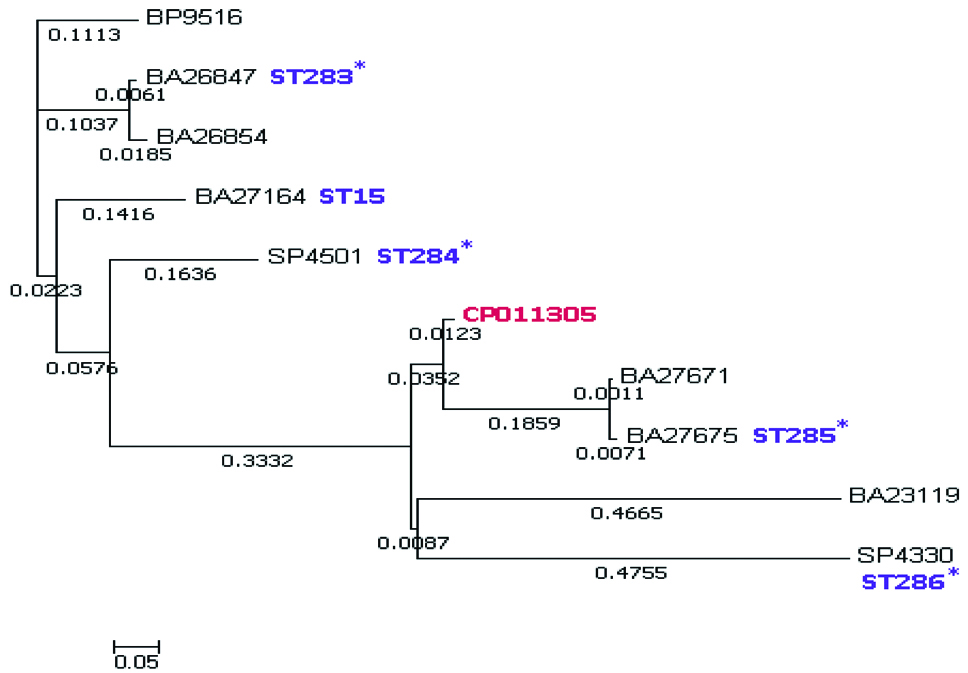
The relation between the AMR phenotypes when compared using cMLST, wgMLST and SNP based typing is given in [Table/Fig-5]. AMR phenotypes showed six different types based on the individual susceptibility profiles. cMLST could not identify any STs. wgMLST identified five STs, of which, four were novel. Whereas, SNP based phylogeny revealed all were different from each other with various root to tip distance values indicating high heterogeneity, except B09516 and B26847, which may be from a single clone source. It can be derived that SNP based method is superior to cMLST and wgMLST. Isolates B26847 and B26854 differ with 0.01 nucleotide substitution per site, however they share the same AMR phenotype, which infers that the nucleotide substitution in the genomes is not related to resistance. Similar results were noted between strains B27671 and B27675.
Comparison of AMR profile with cMLST, wgMLST and SNP based typing methods.
| Isolate ID | AMR Profile(SXT-LEV-CZD-TET-PTZ) | cMLST | wgMLST | SNP phylogenyRoot to tipdistance (nuc. subst./site) |
|---|
| B09516 | CZD-TET-PTZ | - | - | 0.1 |
| B26847 | PTZ | - | ST283 | 0.1 |
| B26854 | PTZ | - | - | 0.11 |
| B27164 | - | - | ST15 | 0.15 |
| S04501 | TET-PTZ | - | ST284 | 0.24 |
| B27671 | CZD-TET-PTZ | - | - | 0.63 |
| B27675 | CZD-TET-PTZ | - | ST285 | 0.64 |
| B23119 | CZD-PTZ | - | - | 0.89 |
| S04330 | TET | - | ST286 | 0.9 |
SXT: Trimethoprim-sulfamethoxazole; LEV: Levofloxacin; CZD: Ceftazidime; TET: Tetracycline; PTZ: Piperacillin/tazobactam; -Not identified; nuc. subst. /site–nucleotide substitution per site
Limitation
The limitation of this study is the small number of isolates tested and the findings need to be validated using a larger number of clinical isolates.
Conclusion
The study reveals that cMLST and wgMLST are not reliable methods for typing of S. maltophilia strains due to frequent recombination events taking place in its genome. Whole genome SNP based phylogeny may help in the discrimination bringing clarity on the clonal relationship. This accurate phylogeny based on SNPs and protein composition may be the molecular typing method of choice for investigating nosocomial spread in the case of S. maltophilia.
SXT: Trimethoprim-sulfamethoxazole; LEV: Levofloxacin; CZD: Ceftazidime; TET: Tetracycline; PTZ: Piperacillin/tazobactam; -Not identified; nuc. subst. /site–nucleotide substitution per site