Bardet-Biedl Syndrome Late Diagnosis with a Great Disability: A Case Report
Oriana Amata1, Enrica Scalisi2, Alessandro Conti3, Giuseppe Umana4, Matteo Cioni5
1 Faculty, Department of Physical Medicine and Rehabilitation, ASST Gaetano Pini-CTO, Milano, Italy.
2 Faculty, Department of Rehabilitation, Humanitas Gradenigo, Torino, Italy.
3 Faculty, Department of Intensive Care Unit, Azienda Ospedaliero-Universitaria “Policlinico Vittorio Emanuele”, Catania, Italy.
4 Faculty, Department of Intensive Care Unit, Azienda Ospedaliero-Universitaria “Policlinico Vittorio Emanuele”, Catania, Italy.
5 Professor, Department of Biomedical and Biotechnological Sciences, University of Catania, Catania, Italy.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Oriana Amata, Via Isocrate, 19 Milano, Italy.
E-mail: amata.oriana@gmail.com
The Bardet-Biedl Syndrome (BSS) is a genetic disease based on autosomal recessive disorder characterised by non-allelic heterogeneity. The prevalence in the European population is only 1 in 160,000 live births. We observed a case of late diagnosis in a patient of 59 years. Different medical specialists, who had seen him before his admission to the hospital, separately treated his different signs and symptoms. The patient was genetically investigated with successfully confirmation of the clinical diagnosis of Bardet-Biedl syndrome was done. This case report underlines the importance of an overview of different clinical signs and symptoms and how different specialties need to collaborate to allow early diagnosis of the diseases.
Autosomal recessive disorder, Genetic disease, Hirschsprung’s disease
Case Report
On 3rd January 2013, a 59-year-old man was admitted in the Intensive Care Unit after surgery for Hirschsprung’s disease.
On physical examination, we observed several abnormalities including: obesity, polydactaly and hypogonadism. The man, weighing 87 kg, the height was 160 cm {Body Mass Index (BMI) was 33.98 kg/m2}, presented postaxial polydactyly of lower limbs (six toes in each foot) [Table/Fig-1] There was facial dysmorphism. He had micropenis and testicles of normal size. Moreover, he had a history of chronic renal failure; his Glomerular Filtration Rate (GFR) was less than 60 mL/min usually.
Post axial polydactyly of lowerlimbs.
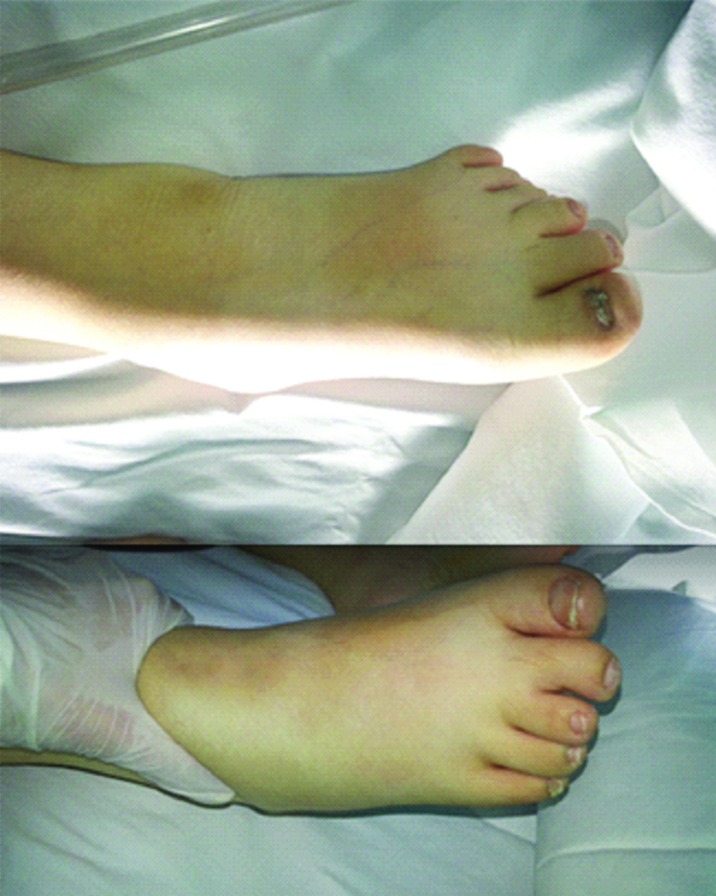
The combination of these clinical features have led us to make a diagnose of BBS in this patient, however nobody in his family has similar impairments.
Laboratory investigations showed that serum blood urea nitrogen and creatinine levels were elevated (80 and 1.4 mg/dL, respectively). Serum testosterone levels were lower than the normal adult male range (4 ng/dL). Meanwhile Luteinizing Hormone (LH) and Follicle Stimulating Hormone (FSH) were 5.7 and 4.9 mIU/mL, respectively.
His family was asked to provide the previous medical documentations. They provided us an abdominal ultrasound, a Computerized Axial Tomography Scan (CAT) and three ophthalmologic evaluations. The abdominal ultrasound exam, of February 2011 and the CAT performed on November 2012, showed polycystic kidneys. Moreover, the ophthalmologic evaluations, including fundoscopy, demonstrated pigmentary retinopathy.
They also showed us some pictures of his childhood and a certificate of mental retardation for Italian government benefits.
To confirm the diagnosis, a blood sample was sent to the CGC Genetics laboratory, with headquarters in Porto. 128 mutations in genes ARL6, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, MKKS, TRIM32 and TTC8, were screened by microarray hybridization. The mutation was confirmed by sequencing (BBS1 ReSeq.:NM 024649.4). The mutation c.951+1G>A in homozygosity was detected in the BBS1 gene. The mutation c.951+1G>A at BBS1 gene is an already known mutation [1]. This result confirmed the clinical diagnosis of BBS and established the molecular aetiology. The genetics laboratory was unable to analyse one of the mutations in gene BBS4 due to technical interference (related on laboratory machine).
The patient and his family signed an informed consent form and also approved publishing of the pictures.
Discussion
This case represents an atypical case of late diagnosis of BBS, in the Intensive Care Unit setting. BBS is a heterogenic and multisystemic chronic disease, discovered in the 1920 by Bardet G [2]. Despite the debilitating conditions, the average age at diagnosis is nine years [3] and the quality of life and disabilities due to clinical features depend of early medical care [4].
The main debilitating conditions include blindness, central obesity, mental retardation, renal failure, reproductive abnormalities.
Each year in the UK, team of geneticist, ophthalmologist, nephrologist, endocrinologist, psychologist, dietitian, speech and language therapist, nurse and a patient support group representative, review patients with BBD. This health care system allows a multidisclipinary management to prevent disabilities resulting from this syndrome [5].
In the present study, a single feature of the enteric nervous system, the Hirschsprung’s Disease (HD), led us to start investigating the patient. Moreover, Hirschsprung’s disease is a very uncommon presentation and very few papers reported the correlation [6,7].
An early diagnosis is essential to prevent and limit the great disability induced by this genetic mutation. It could be useful in planning an appropriate management of clinical manifestations.
Conclusion
The BBS requires the treatments of different clinical features. In fact an early diagnosis of BBS may increase the quality of life.
[1]. Fauser S, Munz M, Besch D, Further support for digenic inheritance in Bardet-Biedl syndromeJournal of Medical Genetics 2003 40(8):e10410.1136/jmg.40.8.e10412920096 [Google Scholar] [CrossRef] [PubMed]
[2]. Bardet G, Sur un syndrome d’obesite congenita leavecpolydactylie et retinite pigmentaire (contribution a l’etudedesformercliniques de l’obesitehypophysaire)Amedee Le Grand 1920 :L470 [Google Scholar]
[3]. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA, New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population surveyJournal of medicalgenetics 1999 36(6):437-46. [Google Scholar]
[4]. O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J, The importance of renal impairment in the natural history of Bardet-Biedl syndromeAmerican Journal Of Kidney Diseases 1996 27(6):776-83.10.1016/S0272-6386(96)90513-2 [Google Scholar] [CrossRef]
[5]. Forsythe E, Kenny J, Bacchelli C, Beales PL, Managing Bardet–Biedl Syndrome-Now and in the FutureFrontiers in Pediatrics 2018 6:2310.3389/fped.2018.0002329487844 [Google Scholar] [CrossRef] [PubMed]
[6]. Lorda-Sanchez I, Ayuso C, Ibanez A, Situs inversus and Hirschsprung disease: two uncommon manifestations in Bardet-Biedl syndromeAm J Med Genet 2000 90:80-81.10.1002/(SICI)1096-8628(20000103)90:1<80::AID-AJMG14<3.0.CO;2-E [Google Scholar] [CrossRef]
[7]. Radetti G, Frick R, Pasquino B, Mengarda G, Savage MO, Hypothalamic-pituitary dysfunction and Hirschsprung’s disease in the Bardet-Biedl syndromeHelvetica Paediatrica Acta 1998 43(3):249-52. [Google Scholar]