A Rare Translocation in a Paediatric Myelodysplastic Syndrome
Rachana Kiran Koppalkar1, Purnima S Rao2, I Sandhya3, Muktha R Pai4
1 Junior Resident, Department of Pathology, A J Institute of Medical Sciences and Research Centre, Mangalore, Karnataka, India.
2 Associate Professor, Department of Pathology, A J Institute of Medical Sciences and Research Centre, Mangalore, Karnataka, India.
3 Associate Professor, Department of Pathology, A J Institute of Medical Sciences and Research Centre, Mangalore, Karnataka, India.
4 Professor, Department of Pathology, A J Institute of Medical Sciences and Research Centre, Mangalore, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Purnima S Rao, 204, Aadheeshwari Apartments, Mahamaya Temple Road, Field Street, Mangalore-575001, Karnataka, India.
E-mail: pursrao@gmail.com
Myelodysplastic Syndromes (MDS) belong to acquired clonal haematologic disorders associated with defective maturation in erythroid, myeloid and megakaryocytic lineages. It rarely affects children and young adults. The common cytogenetic abnormalities in children are monosomy 7, trisomies 8 and 21. Here, authors report a case of MDS in a 15-year-old patient with a rare finding of chromosomal translocation t (6;9) (p23;q34).
Haematology, Haematologic disorders, Multilineage dysplasia
Case Report
A 15-year-old boy presented with on and off neck pain and headache of one-year duration. There was no history suggestive of bleeding tendencies, febrile episodes, loose stools or burning micturition. The child developed swelling of face for which he presented to the hospital. The swelling was persistent without any change. Clinical image could not be obtained as patient/patient party did not give consent for the same. There was no past history of blood dyscrasia or therapies. He had received one unit of blood transfusion in a local hospital the details of which were not available. There was no family history of autoimmune condition. Clinical Examination of the child revealed stable vitals, perioral pallor with absence of organomegaly and lymphadenopathy. Systemic examinations were within normal limits. Clinically on initial assessment anaemia was considered with subsequent investigations to be carried out for unravelling the aetiology.
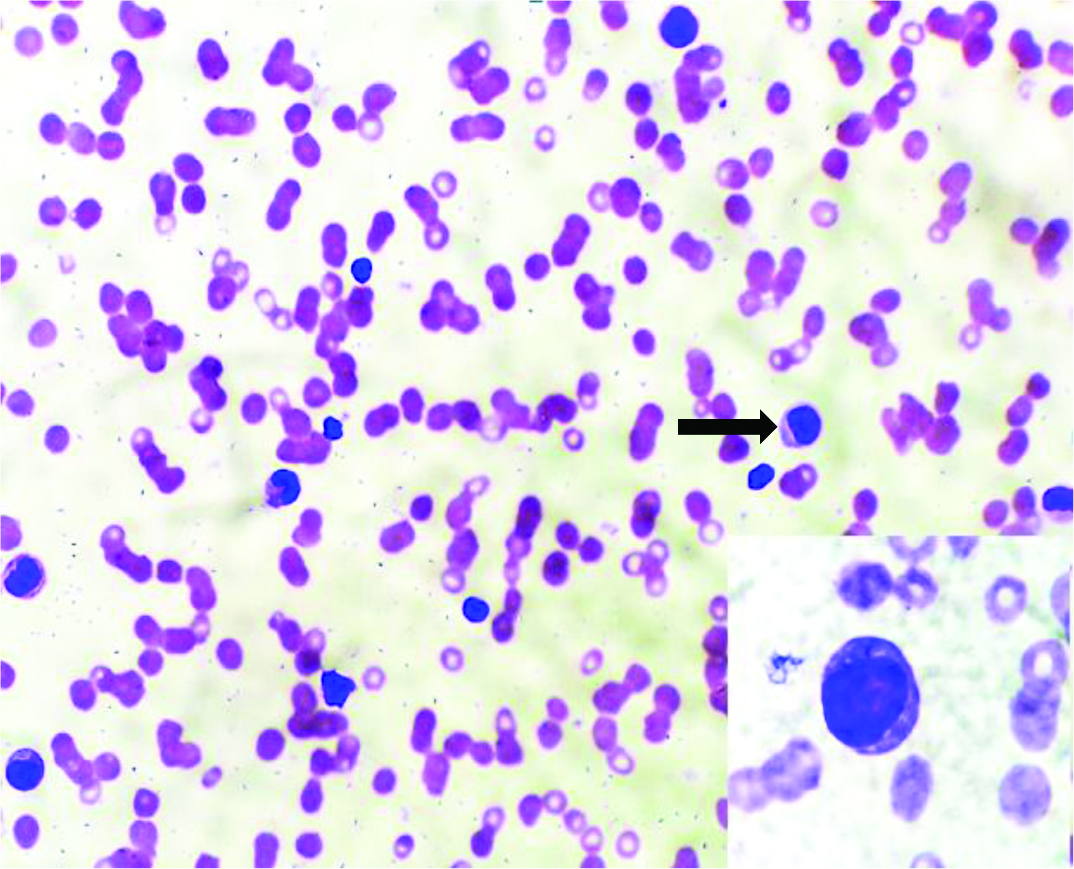
Complete haemogram showed haemoglobin: 9.3 gram/dL, RBC count: 2.36 million/mcL, Total Leukocyte Count (TLC): 11,400/dL, Haematocrit (HCT): 27.8%, Mean Corpuscular Volume (MCV): 117.8+, Mean Corpuscular Haemoglobin (MCH): 39.4+, Mean Corpuscular Haemoglobin Concentration (MCHC): 33.5 and Platelet count: 10,000/dL. Peripheral smear revealed macrocytic red cells, polychromatophils, thrombocytopenia, blasts 6% with monocytosis (16%) [Table/Fig-1]. Reticulocyte count was 1.4%. Monocytosis provides a clue for progression of the disease to acute leukaemia.
Peripheral smear, abnormal cells (Leishmans Stain, x400). Inset shows blast (Leishmans stain, x1000).
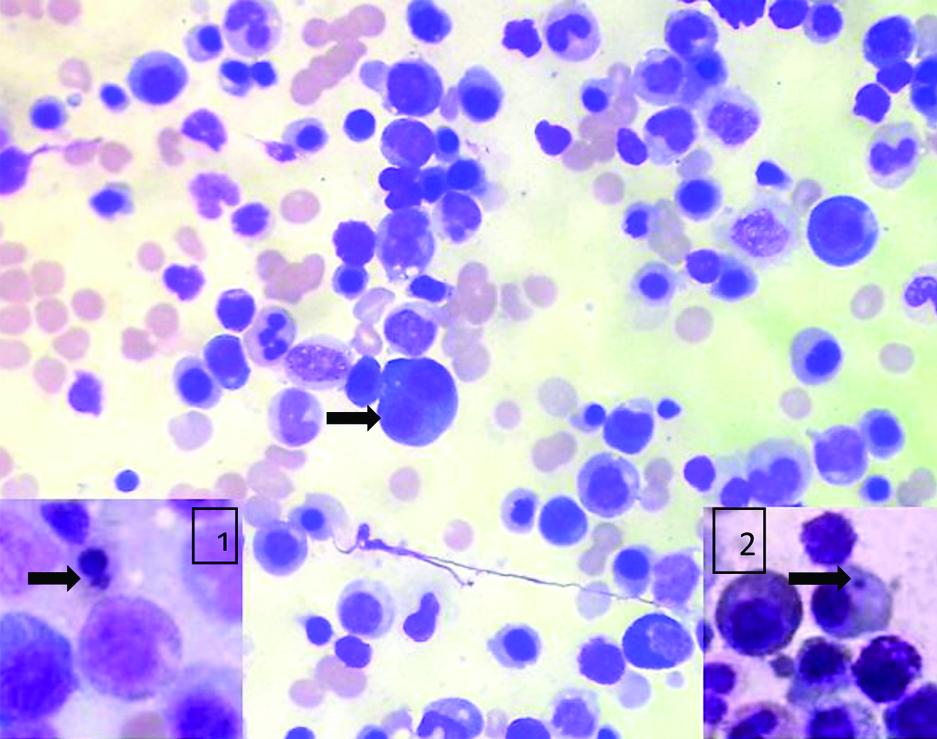
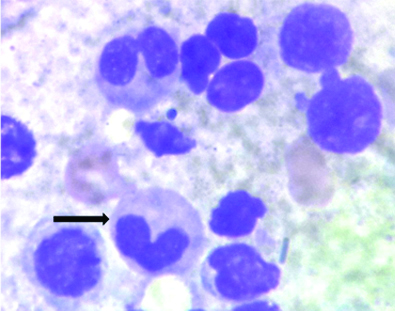
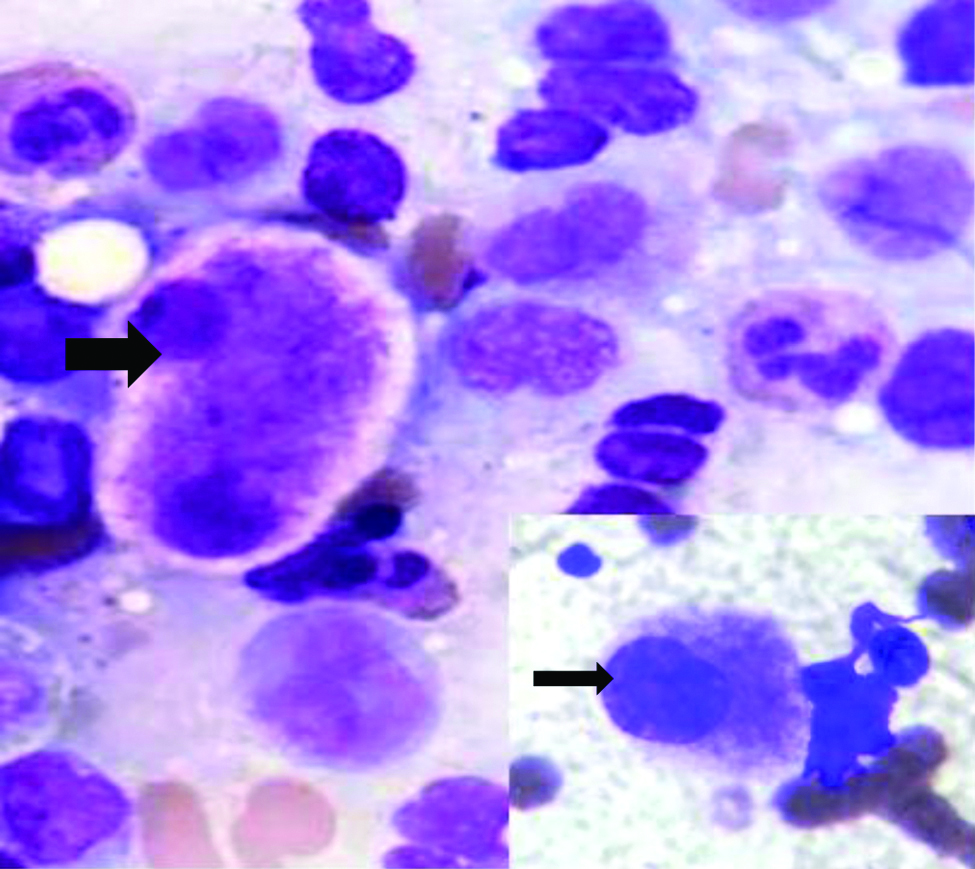
The bone marrow aspiration done was normocellular for age. Erythroid series showed marked dysplasia in the form of nuclear budding, irregular nuclei, megaloblastoid changes, Howell Jolly bodies and multinucleation. No ringed sideroblasts noted. Myeloid series were increased in number with dysplastic forms like large size, pseudo Pelger-Huet nuclei and hypogranulation of neutrophils. Myeloblasts comprised 4% of all nucleated cells. Megakaryocytes were adequate in number with micromegakaryocytes. Bone marrow biopsy exhibited dysplastic megakaryocytes and Grade-3 fibrosis on reticulin stain making it difficult to diagnose blasts. A diagnosis of primary myelodysplastic syndrome was given [Table/Fig-2,3, and 4]. Immunohistochemistry was not done as further analysis by flow cytometry and cytogenetic studies were done. In this case, absence of family history and age at presentation with normal developmental milestones could suggest the possibility of acquired myelodysplastic syndromes, however, genetic testing is necessary for definite diagnosis.
Bone marrow aspirate, erythroid binucleation (MGG, x400); inset 1-erythroid budding (MGG, x1000), inset 2-Howell Jolly body (MGG, x1000).
Bone marrow aspirate, large forms, pseudo pelgerhuet nuclei and hypogranular neutrophils (MGG,x1000).
Bone marrow aspirate, megakaryocyte with abnormally separated nuclear lobes (MGG, x1000); inset-micromegakaryocyte (MGG,x1000).
Bone marrow flow cytometry analysis showed 2.7% immature myeloid precursor cells, 22.8% mature T cells, 1.5% mature B cells and 5.6% monocytes. Flow cytometry dot plot could not be obtained as it was done outside. Fluorescence in-situ hybridisation analysis on interphase cells of the heparinised bone marrow specimen was negative for 5q, 7q, 20q deletions and also for numerical aberrations in chromosome 8. The DNA probes used were LSI 5q EGR1 Spectrum Orange/D5S23 Spectrum Green DC, LSI D7S522 Spectrum Orange/CEP 7 Spectrum Green DC, Vysis directly labelled LSI D20S108 Spectrum Orange and CEP 8 Spectrum Orange. A basic karyotyping carried out revealed clonal structural abnormality in 6p23 in all the cell lines which are associated with bad prognosis. This confirmed the diagnosis of myelodysplastic syndrome with multilineage dysplasia [Table/Fig-5].
GTG banding karyogram of bone marrow cells.
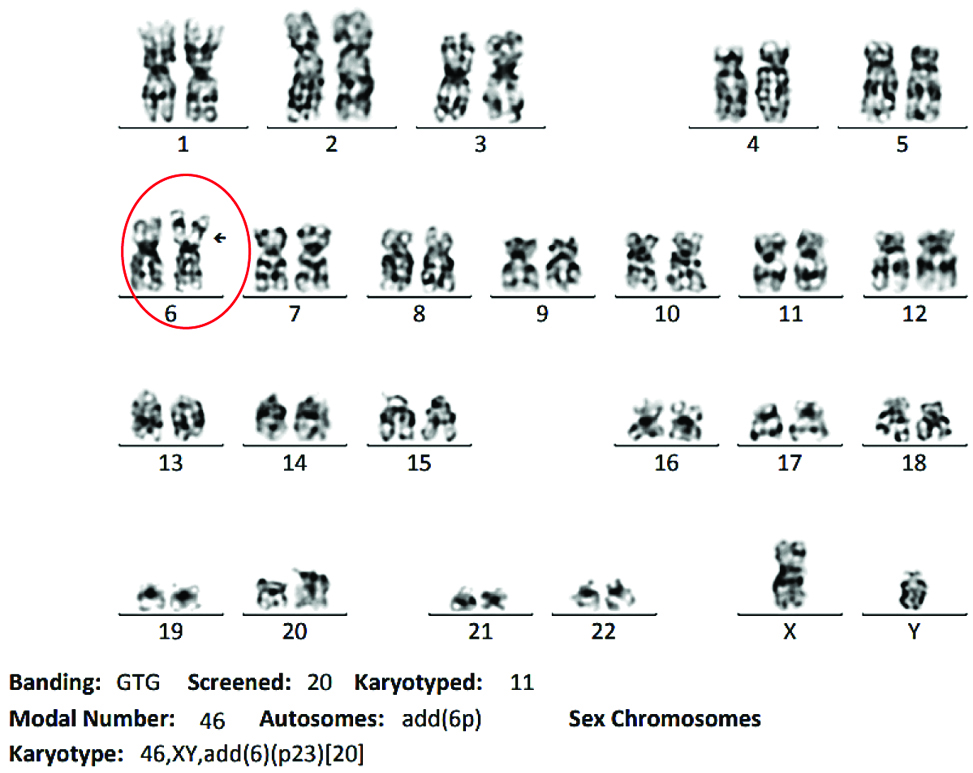
The patient received injection azacytidine 75 mg/m2/day for 7 days every 28 days and was planned and referred for maternal allogeneic transplantation. Following which the patient was lost to follow up.
Discussion
Myelodysplastic syndromes are clonal haematopoietic disorders characterised by ineffective haematopoiesis, bone marrow dysplasia and peripheral cytopenia [1]. MDS accounts for <5% of paediatric haematologic malignancies with an incidence of 1.8-4 cases/million/year worldwide [2] and the incidence in India being 0.65% [3]. Population-based studies estimate the frequency of MDS among childhood haematologic neoplasia to be 6-9% [4]. The median age at disease onset is around 70 years; only about 10% of patients are below the age of 50 [5]. It is unusual for these syndromes to present during childhood and adolescence. The present patient was a rare case of myelodysplastic syndrome in a young boy.
Myelodysplastic Syndrome (MDS) can be a challenging diagnosis, especially when seen in the young. In children, viral infection, metabolic disorders and nutritional deficiencies can mimic MDS. It has historically been categorised as de novo or primary MDS or secondary or therapy-related MDS arising from previous treatment with cytotoxic therapy [6]. The disease biology includes normal clones coexist with abnormal clones. As the disease progresses additional alterations affect the abnormal cells promoting their growth. Abnormal MDS clones harbour altered gene functions that result from single gene mutations, chromosomal abnormalities or epigenetic changes. Recent studies showed increased level of tumour necrosis factor alpha, cytokines such as IL1, IL6, 1L8 have increased apoptosis in MDS clones. In the present case t(6;9)(p23,q34) results in formation of chimeric fusion gene: DEK(6p23) and CAN(9q34). CAN is an oncogene which may be activated by fusion of 3’ end to DEK. Primary MDS is associated with bone marrow failure syndromes such as dyskeratosis congenita, Schwachman Diamond syndrome, Fanconi anaemia [1].
Refractory cytopenia of childhood is the most common subtype of MDS in childhood that account for 50% of all cases [7]. Children usually present with thrombocytopenia in MDS, unlike adults who present with isolated anaemia. In children with MDS, the marrow is usually hypocellular, but in present case the marrow was normocellular.
There were no changes in classification of childhood myelodysplastic syndrome. The Revised World Health Organisation Classification 2016 classifies MDS into MDS with single lineage dysplasia, MDS with ring sideroblasts, MDS with multilineage dysplasia, MDS with excess blasts, MDS with isolated del(5q), MDS unclassifiable and Refractory cytopenia of childhood MDS [7]. Dysplasia in at least 10% of all cells in any one of the lineages, ≥15% ring sideroblasts, 2-19% myeloblasts on blood smears, typical chromosome abnormality. Minimal diagnostic criteria include presence of at least one among the above. One of the less common translocations t(6;9)(p23;q34), although rare in MDS was seen in present case. This translocation was first identified in 1976 and the first paediatric patient was described in 1982 [8]. Children with t(6;9)(p23;q34) had poor prognosis and outcome. Prognostic and risk factors include cytopenias, bone marrow blasts, WHO subtype, karyotype and transfusion requirement. Patients with FLT3 mutation are associated with worse prognosis. Haematopoietic stem cell transplantation in first complete remission improved the 5-year event-free survival of these patients compared with chemotherapy alone [8]. Molecular abnormalities are found in 71% of patients with MDS [9]. Mutations commonly seen in children are genes of the Ras pathway when compared to adults. Germline variants are increasingly recognised that can lead to familial MDS/Acute Myeloid Leukaemia (AML) of which GATA 2 variants have shown to occur in 7% of paediatric primary MDS [10].
The common cytogenetic abnormalities in children are monosomy 7, trisomies 8 and 21; none of which were detected in the present case. Patients with monosomy 7 have a significantly higher probability of progression than do patients with other chromosomal abnormalities or a normal karyotype [11].
It is reported in denovo AML, AML preceded by MDS and AML following chemotherapy [12]. Only two case studies reported in the literature given in [Table/Fig-6]. Higher marrow and peripheral blood blasts were predictive of poor prognosis and shorter overall survival [12]. In a subset of AML, patients showing a similar cytogenetic abnormality can be seen wherein the bone marrow showed increase in monocytes and basophils with associated myelodysplastic features. The two main diagnostic dilemmas are to differentiate MDS with low blast counts from aplastic anaemia and MDS with excess blasts from AML [13].
Case studies with t(6;9)(p23;34) in paediatric MDS patients [8,12].
| Total number of patients | Patients with sole t(6;9)(p23,q34) abnormality | WHO subtype (MDS) | Median age | Gender | Median platelet (/dL) | Additional mutation | Dysplasia |
|---|
| Slovak ML et al., [12] | 31 | 27 | 1 | 13 years | Male | 48,000 | FLT3-11/16 patients | Multilineage dysplasia |
| Sandahl JD et al., [8] | 62 | 47 | 8 | 7.4 years | Male | 76,500 | - | Bilinear dysplasia |
Children with clinical and morphological features of MDS but with cytogenetic features typical of AML-t(8;21)(q22;q22)/t(15;17)(q22;q12) with a score value of 3 as per WHO classification-based Prognostic Scoring System (WPSS)-must be treated as having AML [14]. Advanced MDS patients may benefit from intensive chemotherapy before haematopoietic stem cell transplantation [15].
Conclusion
Paediatric myelodysplastic syndromes are uncommon and may progress to AML. The chemotherapies are not efficacious, bone marrow transplantation is the only curative option. This case was unique as a rare cytogenetic abnormality was recognised; associated with unfavourable outcome and treatment is similar to that of AML. Inspite of performing fluorescence in-situ hybridisation, the final diagnosis was confirmed based onconventional karyotyping alongside clinical and morphological parameters.
[1]. Bannon SA, DiNardo CD, Hereditary predispositions to myelodysplastic syndromeInt J Mol Sci 2016 17(6):83810.3390/ijms1706083827248996 [Google Scholar] [CrossRef] [PubMed]
[2]. Glaubach T, Robinson LJ, Corey SJ, Pediatric myelodysplastic syndromes: They do existJ Pediatr Hematol Oncol 2014 36:1-7.10.1097/MPH.000000000000004624345881 [Google Scholar] [CrossRef] [PubMed]
[3]. Lingegowda A, Kuntegowdenahalli L, Komaranchath A, Devi L, Kumari P, Kamath M, An analysis of the demographic profile, clinical manifestations, investigations and outcome of pediatric myelodysplastic syndrome: A single centre cross sectional studyInt J Cancer Ther Oncol 2015 3(3):33710.14319/ijcto.33.7 [Google Scholar] [CrossRef]
[4]. Stary J, Baumann I, Creutzig U, Harbott J, Michalova K, Charlotte Getting the numbers straight in pediatric MDS: distribution of subtypes after exclusion of down syndromePediatr Blood Cancer 2008 50(2):435-36.10.1002/pbc.2123517455316 [Google Scholar] [CrossRef] [PubMed]
[5]. Kuendgen A, Strupp C, Aivado M, Hildebrandt B, Haas R, Gattermann N, Myelodysplastic syndromes in patients younger than age 50J Clin Oncol 2006 34:5358-65.10.1200/JCO.2006.07.559817088566 [Google Scholar] [CrossRef] [PubMed]
[6]. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, The 2016 revision of the world health organisation (WHO) classification of lymphoid neoplasmsBlood 2016 127(20):2375-90.10.1182/blood-2016-01-64356926980727 [Google Scholar] [CrossRef] [PubMed]
[7]. Swerdlow SH, Campo E, Lee Harris NL, Jaffe ES, Pileri SA, Stein H, Myelodysplastic syndromesIn: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 2017 Revised 4th edLyonIARC:117-118. [Google Scholar]
[8]. Sandahl JD, Coenen EA, Forestier E, Harbott J, Johansson B, Kerndrup G, Adachi S, t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukaemia: an international study of 62 patientsHaematologica 2014 99(5):865-72.10.3324/haematol.2013.09851724441146 [Google Scholar] [CrossRef] [PubMed]
[9]. Germing U, Kobbe G, Haas R, Gattermann N, Myelodysplastic syndromes: diagnosis, prognosis and treatmentDtsch Arztebl Int 2013 110(46):783-90.10.3238/arztebl.2013.078324300826 [Google Scholar] [CrossRef] [PubMed]
[10]. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, Prevalence, clinical characteristics and prognosis of GATA2 related myelodysplastic syndromes in children and adolescentsBlood 2016 127:1387-97.10.1182/blood-2015-09-66993726702063 [Google Scholar] [CrossRef] [PubMed]
[11]. Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR, Refractory anaemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7Blood 2003 102(6):1997-2003.10.1182/blood-2002-11-344412763938 [Google Scholar] [CrossRef] [PubMed]
[12]. Slovak ML, Gundacker H, Bloomfield CD, Dewald G, Appelbaum FR, Larson RA, A retrospective study of 69 patients with t(6;9)(p23;q34) AML emphasizes the need for a prospective multicentre initiative for rare poor prognosis myeloid malignanciesLeukaemia 2006 20:1295-97.10.1038/sj.leu.240423316628187 [Google Scholar] [CrossRef] [PubMed]
[13]. Rau ATK, Shreedhara AK, Kumar S, Myelodysplastic Syndromes in Children: Where are we today?Ochsner J 2012 12:216-20. [Google Scholar]
[14]. Chan GC, Wang WC, Raimondi SC, Behm FG, Krance RA, Chen G, Myelodysplastic syndrome in children: differentiation from acute myeloid leukemia with a low blast countLeukemia 1997 11(2):206-11.10.1038/sj.leu.24005589009082 [Google Scholar] [CrossRef] [PubMed]
[15]. Hasle H, Myelodysplastic and myeloproliferative disorders of childhoodHematol Am Soc Hematol Oncol 2016 2016(1):598-604.10.1182/asheducation-2016.1.59827913534 [Google Scholar] [CrossRef] [PubMed]