In addition to clinical symptoms, clinical history, geographic location, and travel history of the patient, the detection and diagnosis of Leishmaniasis still rely on very old and labour-intensive microscopic validation. Leishmaniasis is an economically orphan disease occurring amongst socially neglected people throughout the world. It is caused by a digenetic protozoan parasite transmitted to vertebrate hosts by the bite of blood-sucking female phlebotomine sand flies [1]. The most severe form of this disease is VL, which is associated with high fatality in absence of proper diagnosis and treatment. More than 70% of cases of VL cases from India are reported from Bihar and the current control measures rely solely on chemotherapy [2]. A robust surveillance system, as well as simple and inexpensive diagnostic kit, is utmost required to control this dreadful parasitic disease [3-5].
Though the microscopic diagnosis remains the gold standard for parasite examination [6], the sensitivity varies from bone marrow smears (65%) to splenic aspirates (>95%) [7]. Furthermore, microscopic evaluation of parasite burden is highly sensitive and mostly relying on the expertise of the microscopist. The induction of strong humoral immune response in active VL patients made it an attractive target for serodiagnosis [8-10]. Often, a simple, rapid immunochromatographic test (rK39) is most frequently used for the diagnosis of VL however; 15-32% of the healthy individual living in the endemic region gave false positive results. This might be due to the activation of the B-cells presents in healthy individuals living in the endemic area, which result in the generation of polyclonal antibody against L. donovani antigens. Furthermore, rK39 illustrate false positive results in relapse cases and the sensitivity varies across the ethnic population (85%-97%) [11]. As the patients suffering from VL reveal a significant increase in the parasite-specific immunoglobulin, thus making it an attractive target for the development of serological tests for VL diagnosis [12]. Several attempts have been made to develop recombinant chimeric antigen expressing immuno-dominant B-cell epitopes of Leishmania spp. for serodiagnosis of VL but none of them showed promising results in the clinical trials or made it to the market [13,14].
Previously, authors had reported the recognition of key antigens (XP_003860226.1 and XP_003861271.1) from Circulating Immune Complexes (CIC) of Leishmania donovani (L. donovani) patients using immunoproteomics approach [15]. Concomitantly, the present study was mainly focused to clone, sequence, express and purify novel proteins and explore their immuno-reactivity using immunoinformatics, immunoblotting and Enzyme-Linked Immunosorbent Assay (ELISA). Also, an attempt was made to evaluate their ability to detect the specific antibody in the sera of active VL cases as well as in other disease backgrounds. Hence, the current study was focused on the precise diagnosis of VL.
Materials and Methods
The present study was designed to evaluate the diagnostic importance of recombinant hypothetical proteins carrying numerous B-cell epitopes. This study includes initial experimental finding which may serve as a basis for the development of the precise and specific VL diagnosis in future. To achieve this target, proteins carrying B-cell epitopes were validated using immunoinformatics approach, and the, proteins under study were cloned, sequenced, expressed and purified. Finally, diagnostic efficacy of the recombinant proteins was evaluated through ELISA.
Study Population
All clinical investigations were conducted at Rajendra Memorial Research Institute of Medical Sciences (RMRIMS), ICMR, Patna, Bihar, India, in the month of September 2017, on human serum sample after getting written, informed consent as per guideline of the Institutional Ethical Committee, registration number: ECR/480/Inst/BR/2014/RR-17 under rule 122DD of the Drug and cosmetic Rule 1945, RMRIMS, Agamkuan, Patna, in accordance with the Helsinki declaration of 1975 which was revised in 2000. Altogether 99 blood samples from human subjects of both sex and all age groups in between 5 to 45 years were studied. For ELISA, sera samples from 22 (VL-BT) along with 11 samples each from Healthy Endemic (HE), Healthy Non-Endemic (HNE), tuberculosis, viral flu, malaria, asthma and filariasis were procured. The sera were stored at -20°C until further use.
Evaluation of Immunogenicity and Conservancy of the Peptide
The potential B- and T-cell epitopes can be determined using immunology focused resources and software, which saves both the time and cost required for laboratory analysis [16,17]. To measure the immunogenicity of the selected peptide, various immunoinformatics algorithms such as BpiPred, ABCpred and EPMLR were used with default threshold value for linear B-cell epitopes prediction [2,18]. BLASTp (http://blast.ncbi.nlm.nih.gov/Blast.cgi) search was performed against the NCBI protein database to retrieve the homologs protein sequences using L. donovani XP_003860226.1 and XP_003861271.1 as query [16]. Epitope conservancy analysis tool from the Immune Epitope Database (IEDB) analysis resource (http://tools.immuneepitope.org/tools/conservancy/iedb_input) was used.
L. Donovani Culture, Isolation of Genomic DNA and Soluble Leishmania Antigen (SLA)
L. donovani culture, genomic DNA isolation and SLA preparation were done according to the protocol mentioned in Singh MK et al., [19].
Amplification and Cloning of L. Donovani rLdhyb and rLdhyc
The rLdhyb and rLdhyc encoding sequences (NCBI Accession no. XP_003860226.1 and NCBI Accession no. XP_003861271.1 respectively) from L. donovani genomic DNA was amplified by PCR. A set of forward (5’TTT GGA TCCATG CTG CGT TTC TGC3’) and reverse (5’AAT AAG CTT TCA GCA CAC CGT CCG3’) primers for were used for rLdhyb and forward (5’-GTC GGA TCC ATG AAG CCG CTA GTG -3’) and a reverse (5’-GTC CTC GAG TCA GCT GTT GTT CTT-3’) primers for rLdhyc. These primers were synthesized commercially (IDT, India). The rLdhyb and rLdhyc gene (XM_003860178.1 and XM_003861223.1) was amplified using the above primers in Thermal cycler (Bio-Rad, USA). Melting temperature (Tm) of forward and reverse primers for rLdhyb was 60.5°C and 59.7°C, and that for rLdhyc were 61.5°C and 58.8°C respectively. The PCR condition consisted of an initial step of denaturation at 95°C for two minutes and followed by 30 cycles of denaturation at 94°C for one minute, with annealing at 58.1°C for one minute, extension at 72°C for one minute and last cycle of final extension at 72°C for 10 minutes. The stock solution was composed with MgCl2, dNTP (Sigma-Aldrich, USA), PCR buffer, forward primer, reverse primer, Taq DNA polymerase (Sigma-Aldrich, USA) and DNA for amplification [Table/Fig-1]. The PCR product was electrophoresed on 1% agarose gel along with 1 Kb ladder (Promega, USA). The amplified product was eluted using the gel extraction kit (Qiagen, Germany). Vector (pET-28a) was isolated from the fresh culture using plasmid isolation kit (Qiagen, Germany). The eluted rLdhyb amplicon of 771 bp was inserted between BamHI and HindIII sites and the rLdhyc amplicon of 1176 bp was inserted in BamHI and XhoI sites of isolated expression vector pET28a (+) (Novagen). Ligation was done using T4 DNA Ligase (Fermentas, Thermo Fisher Scientific, USA). Prior to transfection, chemically competent cells were prepared using 0.1 M CaCl2, followed by heat shock at 42°C. Freshly prepared competent Escherichia coli DH5α cells were transfected with the recombinant plasmid. The transformation was confirmed by colony PCR and restriction double digestion using respective restriction enzymes.
| PCR Components | Stock Solution | Working Solution | Total reaction Volume (25 μL) |
|---|
| MgCl2 | 25 mM | 1 mM | 2.5 μL |
| dNTP | 10 mM | 200 μM | 2.0 μL |
| PCR buffer | 10X | 1X | 2.5 μL |
| Forward Primer | 10 μM | 1 μM | 1 μL |
| Reverse Primer | 10 μM | 1 μM | 1 μL |
| Taq Polymerase | 2.5 u/μL | 2.5 u/100 μL | 0.5 μL |
| DNA | Variable | 500 ng to 1 μg | 2 μL |
| H2O | - | - | 13.5 μL |
DNA Sequence Analysis and Alignment
Gene inserts of rLdhyb and rLdhyc clone were sequenced by Sanger sequencing methods. The cloned genes insert were isolated from vector pET28a with the help of respective restriction enzymes. To verify certainty of the gene cloned in vector, specific forward and reverse primers for rLdhyb and for rLdhyc were used. DNA sequencing was done commercially by Eurofins Genomics. Chromatogram analysis was performed by means of Finch TV 1.4.0 software. Homology searches and sequence alignment were performed using the nucleotide BLAST program http://www.ncbi.nlm.nih.gov/BLAST.
Expression and Purification of rLdhyb and rLdhyc
pET-28a-rLdhyb and pET-28a-rLdhyc construct were isolated from DH5-α and retransformed in competent Escherichia coli BL-21 (DE3, an expression host for recombinant construct). Recombinant proteins were produced by Isopropyl β-D-thiogalactoside (IPTG) induction at 0.8 mM (rLdhyb) and 1 mM (rLdhyc) for 4 hours at 25°C and at 1 mM for overnight at 37°C for rLdhyb and rLdhyc respectively. Expression was confirmed by SDS-PAGE following the protocol of Jamal F et al., [15].
A 600 μL Ni-NTA bead (Qiagen, Germany) in a column was used to purify proteins. The purity of eluted pET-28a-rLdhyb and pET-28a-rLdhyc was analysed by 10% SDS-PAGE (Laemmli, 1970). The Concentration of purified recombinant protein was estimated by the Bradford method using Bovine Serum Albumin (BSA) as standard. SDS-PAGE and western blotting was performed according to the protocol mentioned by Jamal F et al., [15].
ELISA for Detection of rLdhyb and rLdhyc Antibody in VL Patients
For determining most appropriate antigen concentration and antibody dilution, titration was performed at different antigen concentration and antibody serial dilution. Antibody concentration was determined from 1: 50 to 1: 51,000 dilutions. Antigen in form of rLdhyb and rLdhyc was taken in the concentration of 0.5 μg, 1 μg and 1.5 μg per well. ELISA was performed following the protocol mentioned by Jamal F et al., [18].
Statistical Analysis
Statistical analysis was performed using Graph Pad Prism (version 6.0). The lower limits of positivity (cut-off) for the diagnostic antigens were established for optimal sensitivity and specificity. ROC curves were used to determine the ELISA cut-off, sensitivity, specificity and AUC. The cut-off values (dotted line) for negative and positive sample discrimination were calculated using the mean±three times the standard deviation of all negative samples.
Result
In Silico B-Cell Epitopes Prediction
A large number of linear B-cell epitopes were found to be present in recombinant proteins rLdhyb and rLdhyc (XP_003860226.1 and XP_003861271.1 respectively), analysed from different web-servers. [Table/Fig-2a,b], demonstrates numerous B-cell epitopes present in the proteins under study utilising IEDB analysis with the default threshold value set at -0.5. Common best eight epitopes from different servers were selected [Table/Fig-3,4]. The proteins carrying epitopes with good blast score were carried forward for evaluation as a diagnostic candidate. In-depth conservancy analysis of epitopes revealed ~absolute value within species causing the visceralized form of Leishmaniasis [Table/Fig-5,6]. Comparatively B-cell epitopes exhibited meager similarity with Homo sapien [Table/Fig-5,6]. On the other pointer, in silico analysis of whole Ldhyb protein of L. donovani depicted 99% identity with L. infantum, whereas L. braziliensis and L. panamensis shows 68% and 67% identity with 100% query cover. L. major and L. maxicana depicted 92% and 88% identity with 67% query cover. In silico data showed that the target peptide might produce antibodies not only against L. donovani but also against other Leishmania species. Likewise, Ldhyb of L. donovani revealed 99% identity with L. infantum.Ldhyc also revealed 79%-91% identity to other Leishmania spp. with 100% query cover. Ldhyb displayed only 50% identity to Leptomonas spp. with 86-98% query cover. It shows only 32% identity with 40% query cover with Trypanosoma vivax. There was no similarity between Homo sapiens and causative agents of other diseases. Hence, these sequences were conserved among different species of Leishmania.
Linear B-cell epitopes predicted in Ldhyb (XP_003860226.1): (a) and Ldhyc (XP_003861271.1); (b) proteins at default threshold value set at -0.5 using IEDB analysis.
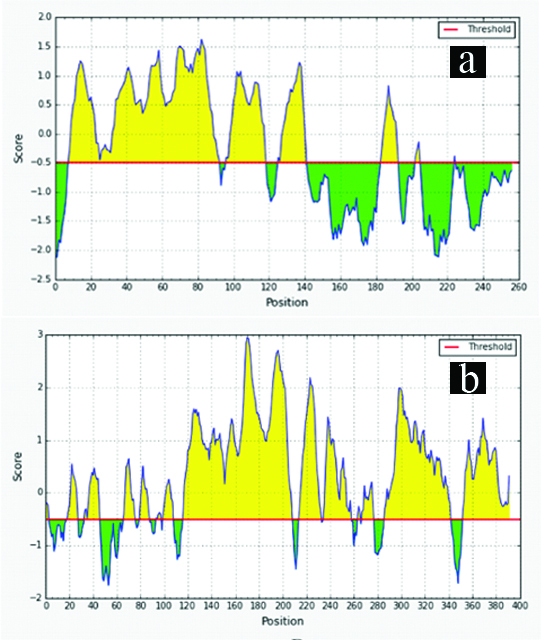
Linear B-cell epitopes prediction of rLdhyb (XP_003860226.1) from different servers.
| Rank | Position | ABCPred (Sequence) | BepiPred | EPMLR |
|---|
| 1 | 37-52 | TGARSTARERRYYTQP | TGARSTARERRY---- | TGARSTARERRYYTQP |
| 2 | 60-75 | TSTIVGRQKSDADAAA | TSTIVGRQKSDADAAA | TSTIVGRQKSDADAAA |
| 3 | 100-115 | GHPRSTTRCRTAHQSP | GHPRSTTRCRTAHQSP | GHPRSTTRCRT----- |
| 7 | 53-68 | LRTPQYGTSTIVGRQK | LRTPQYGTSTIVGRQK | LRTPQYGTSTIVGRQK |
| 9 | 72-87 | DAAATWSGEASSPVAE | DAAATWSGEASSPVAE | DAAATWSGEASSPVAE |
| 9 | 45-60 | ERRYYTQPLRTPQYGT | ERRY-TQPLRTPQYGT | ERRYYTQPLRTPQYGT |
| 13 | 227-242 | REIALHTLCNVFAYVM | REIALH--- | REIALH-LCNVFAYVM |
| 16 | 151-166 | QLRLCTAILAAFYITK | QL…….AFYITK | QLRLCTAILAA----K |
Linear B-cell epitopes prediction of rLdhyc (XP_003861271.1) from different servers.
| Position | ABCPred (Sequence) | BepiPred | EPMLR |
|---|
| 311-326 | TVTKTRPVEVGEPLYS | TVTKTRPVEVGEPLYS | TVTKTRPVEVGEP |
| 123-138 | QRSSDSASGCDARASS | QRSSDSASGCDARASS | QRSSD---------SS |
| 147-162 | LEERHTECFPDPRNDA | LEER-TECFPDPRNDA | LEERHT-PDPRNDA |
| 170-185 | PSSSSSTSASDSVDTA | PSSSSSTSASDSVDTA | P--SSTSASDSVDTA |
| 294-309 | GPASVDPPAAGAITDE | GPASVDPPAAGAITDE | GPASVDPPAAGAITDE |
| 284-299 | TRKRTVPLHFGPASVD | GPASVD | TRKRTVPLHFGPASVD |
| 190-205 | TAAPPSSSPPAAVTPP | TAAPPSSSPPAAVTPP | AAPPSSSPPAAVTPP |
| 129-144 | ASGCDARASSVTPLSP | ASGCDARASSVTPLSP | ------------SSVTPLSP |
Conservancy analysis of predicted B-cell epitopes of rLdhyb.
| Position | Peptide | L. donovani | L. infantum | Homo sapien |
|---|
| 37-52 | TGARSTARERRYYTQP | 100 (100) | 100 (100) | 73 (68) |
| 60-75 | TSTIVGRQKSDADAAA | 100 (100) | 100 (100) | 71 (75) |
| 100-115 | GHPRSTTRCRTAHQSP | 100 (100) | 100 (100) | 75 (50) |
| 53-68 | LRTPQYGTSTIVGRQK | 100 (100) | 94 (100) | 83 (75) |
| 72-87 | DAAATWSGEASSPVAE | 100 (100) | 100 (100) | 58 (75) |
| 45-60 | ERRYYTQPLRTPQYGT | 100 (100) | 100 (100) | 78 (62) |
| 227-242 | REIALHTLCNVFAYVM | 100 (100) | 100 (100) | 67 (93) |
| 151-166 | QLRLCTAILAAFYITK | 100 (100) | 100 (100) | 70 (68) |
*Identity (Query cover)
Conservancy analysis of predicted B-cell epitopes of rLdhyc.
| S No. | Position | Peptide | L. donovani | L. infantum | Homo sapien |
|---|
| 1 | 311-326 | TVTKTRPVEVGEPLYS | 100 (100) | 100 (100) | 88 (50) |
| 2 | 123-138 | QRSSDSASGCDARASS | 100 (100) | 100 (100) | 64 (68) |
| 3 | 147-162 | LEERHTECFPDPRNDA | 100 (100) | 100 (100) | 59 (87) |
| 4 | 170-185 | PSSSSSTSASDSVDTA | 100 (100) | 100 (100) | 80 (56) |
| 5 | 294-309 | GPASVDPPAAGAITDE | 100 (100) | 100 (100) | 89 (56) |
| 6 | 284-299 | TRKRTVPLHFGPASVD | 100 (100) | 100 (100) | 89 (58) |
| 7 | 190-205 | TAAPPSSSPPAAVTPP | 100 (100) | 100 (100) | 60 (75) |
| 8 | 129-144 | ASGCDARASSVTPLSP | 100 (100) | 100 (100) | 64 (87) |
*Identity (Query cover)
Cloning, Expression and Purification of Hypothetical Proteins rLdhyb and rLdhyc
rLdhyb and rLdhyc genes were successfully amplified from genomic DNA isolated from the reference strain of L. donovani (MHOM/IN/83/AG83). Amplification of gene rLdhyb and rLdhyc appeared at ~771bp and ~1.2 Kb region [Table/Fig-7a,8a: Lane 2], which was evaluated in comparison to 1 Kb ladder [Table/Fig-7a,8a: Lane 1]. Double restriction digestion was done with their respective enzymes to confirm cloning [Table/Fig-7b and 8b: Lane 1 and 2]. Western blotting was performed with an anti-His monoclonal antibody, which confirmed the expression and purification of recombinant proteins as a single over-expressed band. Confirmation of purification was shown in [Table/Fig-9] whereas [Table/Fig-7b (Lane 3), 8b (Lane 3)] being broad range (10-250 kDa) molecular weight marker (PureGene, Genetix Brand) respectively.
PCR and confirmation of transformation: a) PCR showing Ldhyb gene amplification. Gene was amplified using a forward 5’TTT GGA TCCATG CTG CGT TTC TGC3’ and reverse 5’AAT AAG CTT TCA GCA CAC CGT CCG3’ primers for rLdhyb using L. donovani genomic DNA, lane 1: Molecular weight marker (1 Kb DNA ladder); lane 2: Amplified PCR gene product; 7b) Double restriction digestion with restriction enzymes to confirm transformation.

PCR and confirmation of transformation: a) PCR showing Ldhyc gene amplification. Gene was amplified using a forward (5’-GTC GGA TCC ATG AAG CCG CTA GTG -3’) and a reverse (5’-GTC CTC GAG TCA GCT GTT GTT CTT-3’) primers for rLdhyc using L. donovani genomic DNA; b) Double restriction digestion with restriction enzymes to confirm insert into vector (lane 2) and only vector (lane 3).

Western blotting showing confirmation of purification with anti-His antibody for rLdhyc, rLdhyb antigenic proteins and broad range MW marker (10-250 kDa) in lane 1, 3 and 2 respectively.

Sequence Analysis of Cloned Genes Inserts using Sanger DNA Sequencing Method
The gene insert in rLdhyb and rLdhyc were sequenced and analysed. The nucleotide sequences of rLdhyb have been deposited in GeneBank under accession number “MH479406” and sequence for rLdhyc was being deposited in GenBank. Both clones were double digested with their corresponding restriction enzyme. Hypothetical protein Ldhyb is an exon region encoded on chromosome number 19; NC_018246.1, encrypted by mRNA (LDBPK_190480) complete cds, comprising 771 bp linear mRNA. Chromatogram acquired by forward primer sequencing revealed 98% identity with 100% query cover (97AGAAGCT-AGCAGC526) with L. donovani strain MHOM/IN/1983/AG83 isolate late passage chromosome 19 accession number CP019526.1 [Table/Fig-10a]. Similarly, reverse primer chromatogram also exhibited 98% identity with 100% query cover (70GGCGCC-TATAGT600) from L. donovani strain CP019526.1 [Table/Fig-10b]. Only 1% gaps were revealed in both ways sequencing owing to nucleotide deletion. Likewise, hypothetical protein Ldhyc is an exon region encoded on chromosome number 24 and encrypted by mRNA (LDBPK_241810) complete cds, comprising 1176 bp linear mRNA. Chromatogram peaks revealed 94% identity with 99% (161CTTGTA-CGAAGG709) and 100% (146ACGCCG-TCATCT887) query cover respectively from forward and reverse primer sequencing respectively, with L. donovani strain Pasteur chromosome number 24 complete sequence; sequence ID: CP022639.1, with 5% Gaps representing nucleotide deletion and insertion [Table/Fig-11a,b]. One and 22 nucleotide insertion site has been identified in cloned gene rLdhyc forward and reverse sequencing respectively. Percentage nucleotide sequence identity between different species has been represented in [Table/Fig-12].
Alignment analysis of cloned gene of rLdhyb showing sequence similarity with L. donovani strain MHOM/IN/1983/AG83 isolate late passage chromosome 19 sequence, Sequence ID: CP019526.1: 10a) DNA sequencing of the cloned gene using the forward primer; 10b) DNA sequencing of the cloned gene using the reverse primer.

Alignment analysis of cloned gene of rLdhyb showing sequence similarity with L. donovani strain pasteur chromosome 24, complete sequence, Sequence ID: CP022639.1. 11a) DNA sequencing of cloned gene using forward primer. 11b) DNA sequencing of cloned gene using reverse primer.

Similarity of cloned sequence with different Leishmania species.
| Leishmania species | Cloned gene (Percentage Identity) |
|---|
| rLdhyb | rLdhyc |
|---|
| Forward Primer Sequencing | Reverse Primer Sequencing | Forward Primer Sequencing | Reverse Primer Sequencing |
|---|
| Leishmania donovani | 98 | 98 | 94 | 94 |
| Leishmania infantum | 97 | 98 | 94 | 94 |
| Leishmania major | 92 | 93 | 89 | 90 |
| Leishmania maxicana | 90 | 89 | - | - |
ELISA
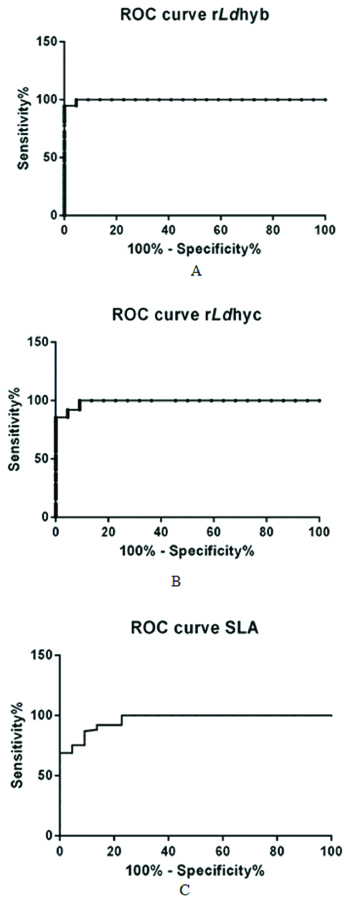
Indirect ELISA was used to determine the reactivity of the recombinant proteins, rLdhyb and rLdhyc against the serological panel. The peptide reactivity was measured with sera from the VL-BT and control groups. Titration curves were performed with ELISA to determine the most appropriate antigen concentration and antibody dilution to be used. One microgram of rLdhyb and rLdhyc were found to be appropriate. Frequency distribution data revealed that ODs for the VL subjects in case of rLdhyb range from 0.39 to as high as 1.019. ODs value for healthy endemic and healthy non-endemic subjects range from 0.11-0.48 and 0.04-0.45, respectively. ODs value for viral flu, leprosy, tuberculosis, malaria, and asthma was 0.21-0.51, 0.07-0.50, 0.047-0.53, 0.15-0.48, 0.17-0.44, respectively, [Table/Fig-13a]. Frequency distribution data revealed that ODs for the VL subjects in case of rLdhyc range from 0.6 to as high as 1.53. ODs value for healthy endemic and healthy non-endemic subjects range from 0.17-0.69 and 0.16-0.67, respectively. ODs value for viral flu, leprosy, tuberculosis, malaria, and asthma were 0.31-0.78, 0.22-0.84, 0.23-0.77, 0.15-0.67, 0.23-0.88 respectively [Table/Fig-13b]. In case of SLA these parameters vary from 0.96-2.00, 0.11-0.48, 0.07-0.4, 0.45-1.16, 0.26-0.97, 0.31-0.84, 0.44-1.12 and 0.38-1.08 [Table/Fig-13c]. The lower limits of positivity (cut-off) for the diagnostic antigens rLdhyb, rLdhyc and SLA were 0.61, 0.95 and 1.31 [Table/Fig-14a-c]. The p-value for each test was <0.0001. Statistical analysis (unpaired t-test) considering VL-BT subject as a test group, whereas healthy and other diseases subjects as control group. The mean, standard deviation, standard error of mean for rLdhyb was calculated to be (0.78, 0.30), (0.15, 0.11), and (0.33, 0.01) respectively [Table/Fig-14a]; for rLdhyc as (1.23, 0.5), (0.24, 0.15) and (0.05, 0.02) respectively [Table/Fig-14b], and for SLA as (1.44, 0.54), (0.38, 0.27) and (0.08, 0.03) respectively [Table/Fig-14c]. Unpaired test was significant with p-value <0.0001. Area under curve for rLdhyb, rLdhyc and SLA were 0.99 and 0.99 and 0.961, with standard error 0.002, 0.007 and 0.019 respectively [Table/Fig-15a-c]. The ELISA revealed the specificity 100%; SpCl (84.56 to 100%); sensitivity 95.4%; Se Cl 95% (87.23-98.57%) for rLdhyb using software-based statistical analysis by GraphPad Prism (version 6.0). For rLdhyc, ELISA revealed the specificity 100%; SpCl (84.56-100%); sensitivity 91%; Se Cl95% (82.16-96.27%), whereas for SLA these data are 100%; SpCl (84.56 to 100%) and 78%; Se Cl95% (67.02-86.58%) respectively. A good diagnostic efficacy of rLdhyb and rLdhyc were 98% and 97%.
ELISA results showing the presence of antibody against rLdhyb, rLdhyc and SLA. Fig 13a, 13b and 13c depicts the frequency distribution graph of antibody for different study groups. The wells were coated with a concentration of 1 μg/well in 100 μL portion of rLdhyb, rLdhyc and SLA. Primary antibody was added in the concentration of 1:100. HRP tagged anti-human secondary antibody was administered in 1:2000 dilution. The ODs was recorded at 450 nm. The ODs value for the VL subjects was two-fold higher than healthy and other disease subjects in case of rLdhyb and rLdhyc.
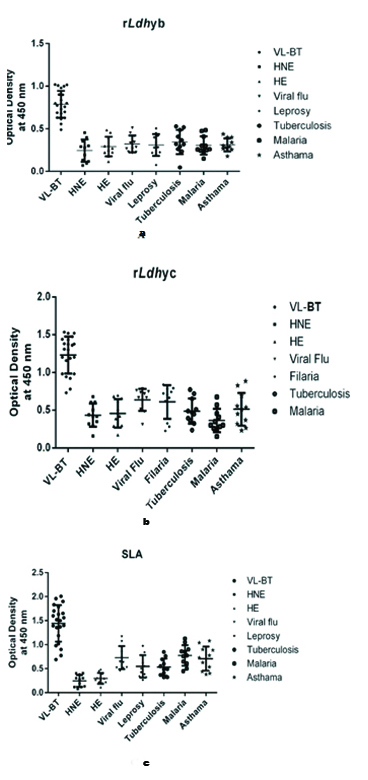
ELISA results representing the lower limit of positivity. Cut-off value for rLdhyb, rLdhyc and SLA were 0.61, 0.95 and 1.31, respectively. The cut-off values (dotted line) for negative and positive sample discrimination were calculated using the mean±three times the standard deviation of all negative samples.

ROC curve. The ELISA revealed the specificity 100% and sensitivity of 95.4% for rLdhyb using software-based statistical analysis by GraphPad Prism (version 6.0). For rLdhyc, ELISA revealed the specificity 100% and a sensitivity of 91%, whereas for SLA these data were 100% and 78% respectively, with the area under curve 0.99, 0.99 and 0.96 respectively, represented by ROC curve (Fig. 15a-c).
Discussion
Labour-intensive practices such as microscopy still endure the mainstay of parasite detection in laboratories. However, over the past few decades, these diagnostic methods have proven to be highly unreliable. Therefore, there is a need for research emphasising the development of more certain tests that do not sacrifice sensitivity and specificity and can be utilised in both clinical settings as well as in poor resource field settings. The arenas of diagnostic parasitology, treatment, and vaccines are undergoing vivid change. In recent years, there has been a tremendous effort to replace the microscopic examination of invasive splenic aspiration, with newer diagnostic methods focusing on serological, molecular, and proteomic approaches.
In the current scenario, the overall sensitivity of available VL diagnostic methods appears highly unsatisfactory. The procedure like splenic bone marrow and the lymph node aspiration are highly invasive and the sensitivity of the parasitological method on aspirates of bone marrow and the lymph node is much lower, and up to 50% of cases of VL are expected to remain untreated if the treatment is conditional on diagnosis confirmation [20]. Upon activation of B-cells specific antibodies are produced which persists comparatively for a long time in patient’s sera and may give false positive results in serodiagnosis [21,22]. Concerning serodiagnosis, it has also been reported that when soluble antigens are used, serological tests are less specific due to cross-reactivity with other parasitic diseases [23]. In the last decades, an increasing number of recombinant protein candidates have been proposed to replace the crude Leishmania antigen (SLA) for the serodiagnosis of leishmaniasis [23]. Major technological advances in recombinant antigens as reagents for the serological diagnosis of VL have led to high sensitivity and specificity of these serological tests. In the existing system, rK39 is the widely used recombinant antigen for VL diagnosis. However, it detects anti-rk39 antibodies in 20-32% of endemic healthy individuals [24]. Further, a report from Sudan has shown the 67% sensitivity of the rK-39 test in immuno-compromised patients [25]. Thus, many efforts have been done to develop more sensitive and specific tests for the detection of VL helping in epidemiology and to control the disease. Hence, there have been the waves of gusto preferring one technique over others.
An endeavour of this study was to ascertain an L. donovani specific antigen which would be highly sensitive and specific. In our earlier study, we identified the B-cell epitopes of L. donovani circulating antigens [15], which specified the presence of B-cell epitopes of two L. donovani hypothetical proteins XP_003860226.1 and XP_003861271.1. This led to the discovery of rLdhyb and rLdhyc proteins as a diagnostic antigen. In the present study employing antigen drawn from blood circulation, hypothetical proteins proved to be very promising with the high degree of specificity and sensitivity. Epitomisation of hypothetical proteins in term of their B-cell epitopes possession exposed a large number of potential B-cell epitopes. Although, majority of B-cell epitopes are conformational or discontinuous; but linear B-cell epitopes are widely accepted for experimental purpose. B-cell epitopes are potentially recognisable by the immune system having the capacity to be bound by antibodies. Towards target, the first phase of the study revealed the epitope nature of targeted proteins. Eight best-matched peptides shortlisted from different servers revealed an excellent B-cell epitopes nature for both rLdhyb and rLdhyc protein [Table/Fig-3,4]. A similar assumption was advocated by Immune Epitope Database (IEDB) analysis Resource, Bpipred, BCpred etc.
Sequence homology analysis of rLdhyb and rLdhyc B-cell epitopes displayed 100% identity with L. infantum [Table/Fig-5,6]. On the other hand, whole protein homology analysis also revealed 99% similarity with L. infantum, mentioned in the result section thus, exhibiting its potential for VL diagnosis. Data also advocated the conservancy of these proteins among different species of Leishmania. Moreover, no any significant similarity could be derived between humans and the causative agent of other diseases resembling in symptoms to VL like tuberculosis, typhoid, malaria etc., henceforth no cross-reactivity and false positive result leading to its absolute specificity.
For the applied investigation hypothetical proteins were successfully cloned, expressed and purified. Further, the logical step was to characterise and prepare recombinant antigens. Western blot result [Table/Fig-9] with anti-His tag monoclonal antibody further authenticated the purified proteins. DNA sequencing data of cloned gene Ldhyb and Ldhyc revealed 98% and 94% identity with L. donovani Gene sequence ID CP019526.1 and CP022639.1 respectively. Cloned gene Ldhyb revealed high similarity with Leishmania spp. hence the likelihood of novel strain of same species. Ldhyc revealed less similarity due to a high percentage of nucleotide deletion and insertion. The results indicated that clones are homologous to the theoretical gene deposited in the Genbank. Also, sequencing result from both the primers revealed the same gene, which data further validated the sequencing results. Sanger sequencing data analysis for different Leishmanial spp. also revealed sequence similarity within species [Table/Fig-12]. To date, several Leishmania antigens have been genetically and antigenically characterised and recombinant technology has been used for the development of novel immunoassays for serological diagnosis of infections [14,26-29]. This study is the novel work where these hypothetical proteins of L. donovani have been characterised and sequenced and reported for the first time.
The study is among the first few works that verify biomarkers from L. donovani hypothetical recombinant proteins for VL patient’s diagnosis. In blood circulation of VL patient, more than 50% of the L. donovani antigens were represented as hypothetical protein [30]. Therefore, a complete hypothetical protein containing a large number of B-cell epitopes might be a better candidate for diagnosis. In silico data was validated by using the recombinant form of proteins. A pertinent and operative approach for the production of large quantities of purified antigens to be used in serological tests is the prokaryotic or eukaryotic systems based on recombinant DNA technology. Thus, providing the best alternative for the serodiagnosis of VL in place of crude antigens used in DAT.
Diagnostic value of immunoinformatics data was authenticated with indirect ELISA, even though it is well known that the short peptides may induce a specific response while large peptide may evoke a non-specific response due to the specificity of antigen. Both the targeted protein under study demonstrated absolute specificity (100%: SpCl (84.56-100)%) on the basis of determined cut-off, due to the presence of enormous epitopes. This excellent specificity is more than any other diagnostics for Kala-azar including rK39 (97%), DAT-FD (Direct Agglutination Test based on freeze-dried-94%), DAT-FDF (Direct Agglutination Test based on freeze-dried using filter paper-94%), Katex (99%), except rK26 which demonstrated 100% value [31]. Although rK39 is associated with high sensitivity, its specificity is a problem in the Indian subcontinent. A large proportion of the healthy population, living in the endemic areas may mimic the signs and symptoms of VL, in case of other diseases like malaria, typhoid fever, tuberculosis, etc., and give rK39 positive result [31]. Thus, there is a crucial need of a diagnostic that could precisely detect active VL.
The ELISA revealed the sensitivity of 95.4%; SeCl95% (87.23-98.57%) for rLdhyb which is better than kinesin based rK26 (22.6%) and antigen detection based KAtex (73%). The sensitivity of rBHUP1 was mentioned as 96.5% however, its low specificity restricted its utility [24]. Earlier, rA2 and BHUP2 proteins were also recognised as a potential target for diagnosis but failed to establish as diagnostics [23,32]. For rLdhyc, ELISA shows sensitivity 91%; SeCl95% (82.16-96.27%). Whereas, for SLA the data is 100%; SpCl (84.56-100%) and 78%; SeCl95% (67.02-86.58%) respectively. Present targeted antigens show better potential than previously documented antigens. The lower sensitivity might be due to non-accessible epitopes region of the cloned protein. Recombinant proteins under study reveal the diagnostic efficacy of 98% and 97% respectively. Hence, further strengthen these antigens’ biomarker property. The recombinant proteins presented a possible applicable approach for VL serological diagnosis since these proteins gave a positive result in the immune assay using many different sera from VL patients and negative results with control groups. Thus, these recombinant proteins with high specificity and sensitivity provide the initial findings for the development of VL specific diagnostics.
Limitation
The studied hypothetical proteins of Leishmania donovani were successfully purified and evidenced their diagnostic potency but in vitro functional characterization of the novel proteins is incumbent. Further large scale validation and authentication of same proteins on different ethnic population is obligatory.
Conclusion
The uniqueness of these L. donovani antigens might be a reason for its high specificity and diagnostic efficacy. As these proteins displayed conservancy of protein sequence within species, these might be used for diagnosing Leishmanial disease other than VL. Moreover, these proteins both in silico and in vitro data established no significant similarity between humans and causative agents of any other diseases. In the present investigation, rLdhyb and rLdhyc antigens revealed an increased ability to discriminate diseases, in comparison to SLA. Though the absolute specificity of rLdhyb and rLdhyc antigens for VL would have made them good diagnostic biomarkers, nevertheless, rLdhyc antigen sensitivity limit its utility. Further evaluation of the different ethnic population may justify their sensitivity. The present findings provide some basic insights for the future development of hypothetical proteins based non-invasive diagnostic tool.