Introduction
Microdeletion syndromes are common genetic disorder characterized by small and variable chromosomal deletion, usually smaller than 5 Mb size (sub-microscopic), in which multiple genes associated with developmental anomalies are involved. The phenotype is mainly due to haploinsufficiency of genes in the critical interval. The common microdeletion syndromes are 22q11.2 (DiGeorge/Velocardiofacial syndrome), 15q11-13 (Prader–Willi/Angelman syndrome), 7q11.23 (Williams-Beuren syndrome), 17p11.2 (Smith Magenis syndrome), 17p13.3 (Miller Dieker syndrome), 17q11.2 (Neurofibromatosis type 1), 22q12.2 (Neurofibromatosis type 2), etc. The 22q11.2 microdeletion syndrome is characterized by hemizygous deletion of <5 Mb size in 22q11.2 region. The prevalence of the syndrome is 1 in 4000 to 6000 live births [1]. Diagnosis of microdeletion syndrome is based on prometaphase banding cytogenetics [2], FISH [3-5], array CGH [6-8], QFPCR [9] with polymorphic micro satellite marker, MLPA [10] and NGS [11,12].
Mosaicism is defined as the presence of two or more population of cells with different genotypes derived from single zygote. Mosaicism in microdeletion syndrome is commonly viewed as rare and reported infrequently [13-15]. Interphase FISH analysis is the prime method for diagnosis of mosaicism, although MLPA, microarray and NGS can also be used to study mosaicism. The FISH analysis has the ability to determine the presence of a particular DNA sequence in cells (qualitative) along with information on numbers (quantitative) besides their anatomic/physical location in cell to cell basis i.e., cells type, number, etc.,. Interphase FISH can diagnose mosaicism rapidly and reliably as this can analyse large number of cells individually and even low level of mosaicism [14,16]. Diagnosis of 22q11.2 microdeletion mosaicism through FISH on prenatal samples [17], on peripheral blood lymphocytes [14,16,18,19] and on cardiac tissue [19] have been reported earlier [Table/Fig-1] [13,14,16,17,20-35]. SNP microarray is another good method to screen clinically suspected microdeletion cases [8], as this single test provides information on genome at high resolution on aneuploidy, polyploidy, microdeletions, microduplications, parental inheritance/uniparental disomy, but often may fail to detect low level mosaicism, if not carefully analysed. The present study was conducted to observe mosaicism, which is one of the leading mechanisms for phenotypic heterogeneity associated with the 22q11.2 microdeletion syndrome.
List of publications citing mosaicism in various microdeletion syndromes [13,14,16,17,20-35].
| Microdeletion | Tissue examined | Method | Result | Reference |
|---|
| 3q29 | Blood nucleated cells | QF PCR | 40% deleted cells (mosaicism) in carrier father | [26] |
| 9q34.3 | Blood nucleated cells and fibroblast cells in family 1; buccal cells in family 2 | FISH | Family 1 carrier mother had less deleted cells in fibroblast than bloodFamily 2 carrier mother had 80% buccal cells with deletion | [24] |
| 12q24.31-q24.33 | Interphase lymphocytes | FISH | 44.5% normal, 30% hemizygous deletion and 25.5% homozygous deletion | [27] |
| 15q11-13 | Metaphase lymphocytes | FISH | 40% deleted cell lines (case report) | [28] |
| 15q11-13 | Metaphase lymphocytes | FISH | 15% deleted cells | [16] |
| 15q11-13 | Metaphase lymphocytes | FISH | 58% deleted metaphase cells (lymphocytes) | [29] |
| 17p11.2 to17p12 | Interphase lymphocytes | FISH | 12% deleted cells(case report) | [25] |
| 17q11.2 | Interphase lymphocytes | FISH | 40% (8/20) cases had somatic mosaicism; deleted cell line was 91-100% in blood cells but 51-80% in buccal and skin cells | [21] |
| 17q11.2 | Metaphase lymphocytes | FISH | 22.2% (2/9) cases had somatic mosaicism | [30] |
| 17q11.2 | Blood WBC and DNA (derived from peripheral blood) | FISH, MLPA, QF PCR | 44% in NF1 type 2 & 25% in NF1 atypicalOverall 9.6% (14/146) had mosaicism (deleted cells were 50% to 97%) | [20] |
| 17q11.2 | Metaphase lymphocytes and skin fibroblast cells | FISH | Mother had deletion in 70% lymphocytes and 15% fibroblast metaphase cells | [23] |
| 17q21.31 | Interphase and metaphase lymphocytes and buccal cells | FISH | Family 1 carrier mother had 8% cells with deletion in buccal cells and Family 2 carrier mother had 7% cells with deletion in buccal cells but 3% in interphase and metaphase lymphocytes | [22] |
| 20p12 | Blood WBC | FISH | Mother carrier had deletion in 50% of peripheral blood cells | [31] |
| 21q22 | Amniocytes | FISH | 14-97% deletions with different probes (complex mosaicism) | [32] |
| 22q11.2 | Interphase lymphocytes | FISH | 7% cells with deletion in carrier mother with 2 affected sons | [33] |
| 22q11.2 | Interphase amniocytes | FISH | 61% cells deleted | [17] |
| 22q11.2 | Metaphase and interphase lymphocytes and fibroblasts | FISH | 55% (11/20) cells with deletion and 45% (9/20) cells with duplication in metaphase lymphocytes85.7% (18/21) metaphase fibroblasts and 40/50 (80%) interphase fibroblast with deletion | [13] |
| 22q11.2 | Interphase lymphocytes, buccal cells and urinary cells | FISH | 78-89% cells deleted in case 1 in various cell type12-15% cells deleted in case 2 in various cell type | [14] |
| 22q11.2 | Interphase lymphocytes | FISH | 70% lymphocytes with deletion | [34] |
| 22q11.2 | Interphase lymphocytes | FISH and microarray | 66% cells deleted in affected fetus | [13] |
| 22q13.3 | Blood WBC | FISH | 6% deletion in carrier mother with 2 affected children | [35] |
Materials and Methods
This study was conducted in the Department of Reproductive Biology, All India Institute of Medical Sciences (AIIMS), New Delhi, India. From January 2005 to July 2017, 257 cases of clinically suspected 22q11.2 microdeletion syndrome were evaluated using FISH method [3]. The study was approved by the Institutional Human Ethics Committee (Ref. No. IEC/NP-93/11.4.14 & A-21:12/08/2005). Procedure was in accordance with the Helsinki declaration. All cases were referred from various parts of North India and mostly included referred cases with normal karyotype, excluding very sick neonates. All 257 cases (excluding very sick neonates) underwent clinical evaluation as per guideline [36] before undergoing specific FISH test for 22q11.2 microdeletion. There were 164 males and 93 females between ages 5 days to 15 years. EDTA and heparinized blood samples were collected from most patients and their parents after obtaining written consent for the study. FISH study was carried out on both interphase and metaphase cells using non-commercial FISH probe [3,4]. PAC (P1-based artificial chromosomes) clone (RP5-882J5/dJ882J5 for 22q11.2) was obtained from European Resource Centre for Molecular Cytogenetics, University of Bari, Italy (http://www.biologia.uniba.it/rmc/; courtesy Professor Mariano Rocchi) for the study. The clone was received as bacterial LB agar stab culture, processed in house and used as directly flurochrome labelled FISH probe [5,14]. The origin of this clone was in Pieter de Jong library as RPCI-5: Human (M) PAC library. An RPCI Human PAC segment cloning vector was pCYPAC2, average insert size was 115 kb and genomic coverage was 6X. Standard cytogenetic techniques were used to obtain metaphase lymphocytes chromosomes. In short blood microculture was planted in the presence of mitogen and after 70 hours, a mitotic inhibitor was added to stop mitosis (arrest cells in metaphase) followed by hypotonic treatment and fixation in methanol acetic acid solution. For interphase cells preparation about 100μl blood was processed into a 1.5ml micro centrifuge tube containing 400μl PBS (phosphate buffered saline, pH7.4, Sigma), mixed well and centrifuged at 5000 rpm for 5 minutes. The supernatant was discarded and this washing procedure was repeated three times followed by adding 400μl hypotonic solution (50mMol KCl) into the pellet and incubated for 20 minutes before adding fixative (3:1 methanol and acetic acid). Approximately 15-20μl of cell suspension was used to make slide. Probes and cell DNA were denatured together for 3 minutes at 75°C and incubated at 37°C in a moist chamber for overnight. Post hybridization washing was done using NP40 solution. The slides were counterstained and mounted using antifade containing DAPI and screened under fluorescent microscope using appropriate filter sets. Probe hybridization efficiency (sensitivity and specificity) was evaluated in control metaphase spread with each FISH experiment. We found no cross hybridization and the probe hybridized specifically on chromosome 22q. We have also evaluated FISH probe hemizygous deletion prevalence in various cells in normal individual [Table/Fig-2]. An evaluation target of at least 500 interphase nuclei (50 interphase cells as per ACMG FISH standard and guideline) [37] and 10 metaphase nuclei (more in case of low level mosaicism) were attempted in every case. Presence of both one and two signals in metaphase (at least 5%) and interphase (at least 10%) cells were observed, the case was considered to have mosaicism [14]. In absence of metaphase chromosome, mosaicism was considered when at least 35% interphase cells display one or two signal/s in any type of cells (blood, buccal or urinary). Interphase FISH was carried out on buccal (ectodermal origin) and urinary cells (endodermal origin) as described before [38] to find out whether mosaicism restricted to blood or generalized. Diagnosis of mosaicism was assigned after examining various factors viz., clinical features, deletions in metaphase chromosome (>5%) and/or deletions in interphase cells.
The 22q11.2 microdeletion probes hybridization efficiency in control (normal) lymphocytes (post cell culture), nucleated blood (granulocytes & mononuclear; without cell culture), buccal (squamous cells) & urinary (uroepithelial cells) cells (interphase).
| 22q11.2 FISH probe(RP5-882J5) | Number of controls studied | Dizygous/2 signals | Hemizygous/1 signal (%) | Others/3-4 signals |
|---|
| Peripheral blood lymphocytes(interphase post culture) | | | | |
| FISH result | 131 | 48933 | 832 (1.66%) | 127 |
| Observed hemizygous deletion in normal individual | | | 1.7% cells | |
| Peripheral blood nucleated cells (WBC)(interphase uncultured) | | | | |
| FISH result | 5 | 488 | 19 (3.6%) | 9 |
| Observed hemizygous deletion in normal individual | | | 3.6% cells | |
| Buccal ells | | | | |
| FISH result | 5 | 418 | 20 (4.5%) | 8 |
| Observed hemizygous deletion in normal individual | | | 4.5% cells | |
| Urinary cells | | | | |
| FISH result | 5 | 277 | 31 (9.8%) | 7 |
| Observed hemizygous deletion in normal individual | | | 9.8% cells | |
Results
In this study the prevalence of mosaicism in 22q11.2 microdeletion has been assessed (DiGeorge) syndrome by FISH and importance of examining various tissues to detect mosaicism. A total of 257 cases of clinically suspected 22q11.2 microdeletion syndrome were studied by FISH using 22q11.2 probe (RP5-882J5) as per guideline for 22q11.2 microdeletion FISH test [36]. The 22q11.2 FISH probe hybridization efficiency in normal control was observed as two signals (dizygous) in about 98% nuclei in interphase lymphocytes (cultured), about 96% nuclei in nucleated blood cells (uncultured), about 95% nuclei in buccal cells and about 90% nuclei of urinary cells [Table/Fig-2]. Hemizygous deletion in normal individual was observed as about 2% in interphase lymphocyte (cultured) and without culture in about 3.6% in blood nucleated cells, about 4.5% in buccal cells and 9.8% in urinary cells, respectively. This indicates normally with this probe FISH on interphase cells one may expect one signal/one copy at maximum in about 10% cells. The reason for background (procedural) low level hemizygous deletion (1 signal) is due to various factors like probe hybridization efficiency, superimposition of signals, chromosome segregation error, etc.,. The 22q11.2 microdeletion was identified in 39 (15.2%) cases. Mosaicism was detected in 11 (28.2%) cases [Table/Fig-3,4,5,6 and 7]. Proportion of deleted cell in various tissues was 10-90% in interphase cells [Table/Fig-7]. Clinical comparison depending upon deletion load (<35%, 35-65% &>65% deleted cells) in mosaic cases is presented in [Table/Fig-8]. No co-relation was detected between deleted cell concentration and phenotypic presentations [Table/Fig-8]. Neither 22q11.2 microdeletion nor even low level of mosaicism was observed in parents of the analysed cases.
Summary of FISH results in 22q11.2 microdeletion syndromes.
| Parameters | 22q11.2 Microdeletion | Remarks |
|---|
| FISH probe details | RP5-882J5 | Non-commercial probes |
| Clinically suspected cases tested by FISH | 257 | Adequate number of cases evaluated |
| FISH deletion positive cases | 39 (15.2%) | Low detection rate |
| Pure/non-mosaic deletion in peripheral blood lymphocytes (mesodermal origin) | 28 (71.8%) | Pure/non-mosaic deletion cases 71.8% |
| Mosaic deletion | 11 (28.2%) | Mosaic deletion 28.2% |
| Remarks | High frequency of mosaicism | |
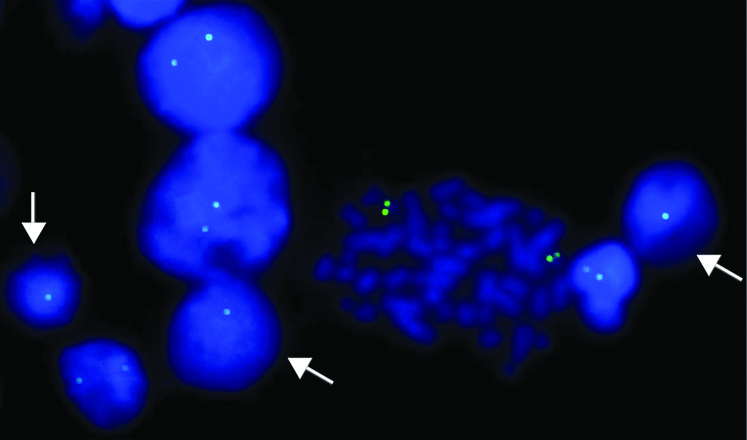
FISH image using 22q11.2 locus specific probe (labeled with FITC) on lymphocyte culture cells showing dizygous/normal (two green signals) cells (one metaphase and four interphase cells) and hemizygous/deleted (one green signal) cells (arrow).
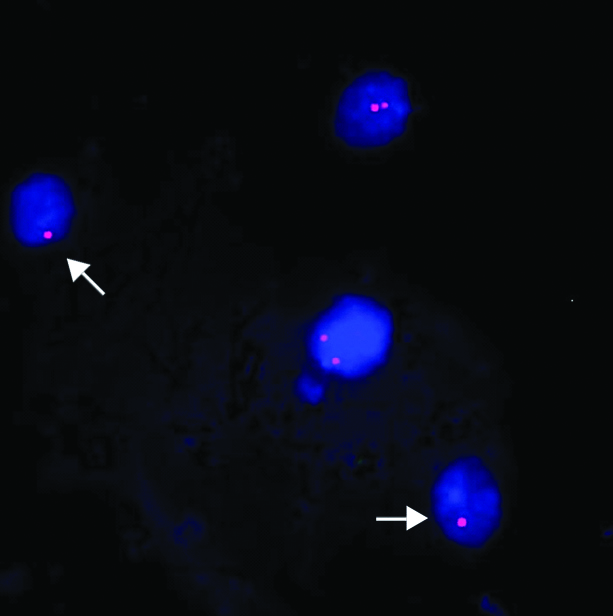
FISH image using 22q11.2 locus specific probe (labelled with Cy3) on buccal cells showing dizygous/normal (two red signals) cells and hemizygous/deleted (one red signal) cells (arrow).
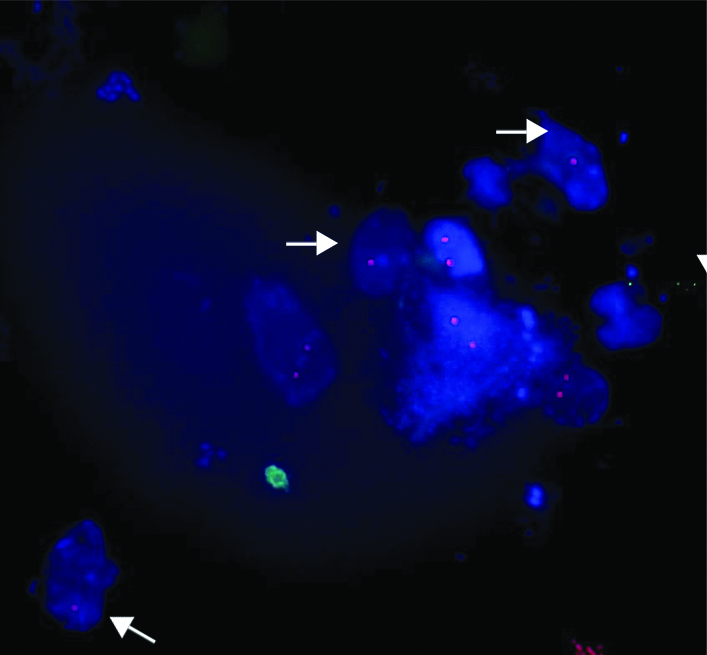
FISH image using 22q11.2 locus specific probe (labelled with Cy3) on urinary cells showing dizygous/normal (two red signals) cells and hemizygous/deleted (one red signal) cells (arrow).
Interphase (blood, urine and buccal cells) and metaphase (blood) FISH result in mosaic 22q11.2 microdeletions.
| CasesAge/Sex | Metaphase FISH in blood lymphocytes | Interphase FISH in blood cells | Interphase FISH in buccal cells | Interphase FISH in urine cells | Remarks |
|---|
| 1½ months/ Malevery sick (in ICU; expired), HC, TH, CL/ CP, Cyanosis, F/H of CHD, etc., | 1 copy in 3 metaphase cells (27.3%),2 copies in 8 metaphase cells (72.7%) | 1 copy in 170 cells (26.5%),2 copies in 470 cells (73.5%) | Not done | Not done | About 27% metaphase have deletion |
| 10 months/ MaleTOF, Dys, NPI, LSF, F/H of CHD, etc., | 1 copy in 3 metaphase cells (27.3%),2 copies in 8 metaphase cells (72.7%) | 1 copy in 228 cells (31%),2 copies in 500 cells (68.7%) | 1 copy in 51 cells (94%),2 copies in 3 cells (6%) | 1 copy in 25 cells (92.6%),2 copies in 2 cells (7.4%) | >27% metaphase has deletion; Buccal and urinary cells show over 90% deletion whereas blood cells show approximately 30% deletion |
| 1 year/ MaleTOF, Dys, DD, CP, LSF, etc., | 1 copy in 7 metaphase cells (77.8%),2 copies in 2 metaphases (22.2%) | 1 copy in 500 cells (86%), 2 copies in 84 cells (14%) | Not done | Not done | 14% metaphase/ interphase cells have no deletion |
| 1½ years/ MaleCHD (VSD), Dys, DD, LSF, etc., | Blood culture not attempted due to EDTA blood | 1 copy in 319 cells (46.6%); 2 copies in 365 cells (53.4%) | Not done | Not done | 46% deletion in interphase cells |
| 1½ years/ MaleTOF, Dys, DD, NPI, LSF, Trigonocephaly, etc., | 1 copy in 6 metaphase cells (15%),2 copies in 34 metaphase cells (85%) | 1 copy in 121 cells (11.6%),2 copies in 925 cells (88.4%) | 1 copy in 8 cells (13.8%),2 copies in 50 cells (86%) | 1 copy in 7 cells (14.9%),2 copies in 40 cells (85%) | 15% metaphase have deletion; 12-15% of interphase cells have deletion |
| 2 years/ MaleTOF, Dys, DD, polydactyly, MCK, etc., | 1 copy in 4 metaphase cells (40%),2 copies in 6 metaphase cells (60%) | 1 copy in 183 cells (54%),2 copies in 155 cells (46%) | Not done | Not done | 43% metaphase have deletion |
| 2 years 10 months/ MaleTOF, Dys, DD, HAP, LSF, etc., | 1 copy in 50 metaphase cells (94.3%),2 copies in 3 metaphase cells (5.6%) | 1 copy in 2900 cells (85.3%),2 copies in 497 cells (14.6%) | 1copy in 14 cells (78%),2 copies in 4 cells (22%) | 1 copy in 8 cells (89%),2 copies in 1 cell (11%) | >5% metaphase have no deletion; 11-22% interphase cells without deletion |
| 3 years/ MaleTOF, Dys, DD, NPI, LSF, etc., | Blood culture failed | 1 copy in 500 cells (47.6%),2 copies in 550 cells (52%) | 1 copy in 18 cells (72%),2 copies in 7 cells (28%) | 1 copy in 12 cells (70.5%),2 copies in 5 cells (29.5%) | 47-72% interphase cells have deletion |
| 15 years/ MaleTOF, Dys, HC, SD, tall, etc., | 1 copy in 2 metaphase cells (25%),2 copies in 6 metaphase cells (75%) | 1 copy in 150 cells (26%), 2 copies in 430 cells (74%) | 1 copy in 8 cells (10%),2 copies in 70 cells (90%) | 1 copy in 4 cells (12.5%),2 copies in 28 cells (87.5%) | 25% metaphase have deletionLow level (10-26%) of interphase cells have deletion |
| 1 month/ FemaleCHD (VSD, PDA, etc.,), Dys, HC, LSF, etc., | 1 copy in 4 metaphase cells (40%);2 copies in 6 metaphase cells (60%) | 1 copy in 160 cells (49%), 2 copies in 164 cells (51%) | 1 copy in 11 cells (27.5%),2 copies in 29 cells (52.5%) | 1 copy in 2 cells (28.5%),2 copies in 5 cells (71.5%) | 40% metaphase have deletion27-49% interphase cells have deletion |
| 3 years/ FemaleTOF, Dys, DD, HAP, LSF, etc., | 1 copy in 5 metaphases (50%),2 copies in 5 metaphase cells (50%) | 1 copy in 260 cells (51%), 2 copies in 252 cells (49%) | Not done | Not done | 50% metaphase lymphocytes have deletion |
TOF: Tetralogy of fallot; Dys: Dysmorphism; DD: Developmental delay; HAP: High arched palate; LSF: Long slender fingers; CP: Cleft palate; NPI: Nasopharyngeal insufficiency; MCK: Multicystic kidney; CHD: Congenital heart defect; VSD: Ventricular septal defect; PDA: Patent ductus arteriosus; HC: Hypocalcemia; F/H: Family history; ICU: Intensive care unit; TH: Thymic hypoplasia; CL: Cleft lip; SD: Skeletal deformity
Discussion
In this study 11 mosaic cases, have been detected among 39 cases of 22q11.2 microdeletion syndrome, giving a frequency of 28.2% mosaicism. We have also observed low level (<35%) of deleted cell lines in over 36% (4/11) cases. Until now, mosaicism in common microdeletion syndromes was reported as case reports or few case series [Table/Fig-1] and considered as rare [13-17]. This study has shown comparatively high incidence (28.2%) of mosaicism in 22q11.2 microdeletion syndrome. This study, therefore demonstrates that mosaicism represents an important context in the investigation of 22q11.2 microdeletion syndrome. High frequency of mosaicism is also reported with NF1 (Neurofibromatosis 1) type 2 (44%) and NF1 atypical (25%) microdeletion syndromes [20]. Somatic mosaicism i.e., variation in deletions in different somatic tissues [Table/Fig-7] viz., blood nucleated cells (mesodermal origin), buccal cells (ectodermal origin) and urinary cells (endodermal origin) were also observed. This is also reported in NF1 [20,21] as well as other microdeletion syndromes [13,22-24]. In addition, we have observed wide variations in deleted cell concentration in different type of tissues using interphase FISH. We observed near similar concentration in all 3 germ layers but in some cases, there were wide variations between blood (mesodermal) and others tissues (ectodermal and endodermal). Hence, the use of interphase FISH on blood and other cell types is advisable as prime detection technique for mosaicism. This is very important during screening parents of an affected child as often one of the parents may have low level of mosaicism in some or all tissues [26,27] or gonad that may cause recurrence (as high as 50%) [39]. In this situation, FISH can analyse large number of cells quickly and can help to determine carrier state of parents, thus helps in accurate reproductive genetic counselling although no carrier parents were found in this study. Gonadal mosaicism in father can also be determined easily from sperm FISH. However, gonadal mosaicism in mother is difficult to assess as this requires either polar body or oocyte FISH (single cell and invasive/assisted reproductive technology). Low-level mosaicisms (7-35% of abnormal cells) was reported with various microdeletion syndromes [14,16]. However, FISH provides information only on targets and does not allow a comprehensive evaluation of atypical deletions outside the region of FISH probe [8].
In mosaicism individuals have two or more cell lines with genetic differences. These cell lines are derived from single zygote but shows variation due to post-zygotic mutational events. The mechanism of mosaicism involves mitotic error during cell division through non-disjunction (for chromosome numbers) or anaphase lag (for chromosome numbers) or structural abnormalities (for translocations, deletions, duplications, marker or ring chromosomes), including Non-Allelic Homologous Recombination (NAHR). Most copy number variations (microdeletion and microduplication) result from meiotic and/or mitotic NAHR. Somatic NAHR is mediated by intra-chromosomal exchange during mitosis [40] and somatic NAHR may be responsible for the occurrence of somatic mosaicism. Cytoplasmic abnormalities such as impaired cytoskeleton and spindle malfunction or Low Copy Repeat (LCR) sequences influence abnormal DNA segregation (copy number variation) or cell division (aneuploidy) and thus leads to mosaicism [7]. Appearance of rescue attempt at a particular time during embryogenesis determines the proportion of affected cells. Rescue of mutated cells through the process of recombination after fertilization results in mosaicism and may confer advantage in survival. Mosaicism is considered as an important mechanism of phenotypic heterogeneity in microdeletion syndrome [25,41]. However, in this study we did not find any co-relation between load of deleted cells and severity of disease [Table/Fig-8]. We have found classical phenotype with low-grade mosaicism (<35% cells with deletion) as well as milder phenotype with high-grade mosaicism (>65% cells with deletion). Similarly, other researchers also observed classical phenotype with mosaicism [42], even with low level of mosaicism [43]. This difference could be due to tissue mosaicism; often non-blood cells have more deleted cells than blood cells and vice versa as seen with some of our cases [Table/Fig-7]. Reason of phenotypic variability is complex and depends on many factors, including deletion size [7], associated CNVs (copy number variation), tissue mosaicism [14,16], parental origin of deletion, and epigenetic changes, however further studies are required in this field.
Summary of clinical presentation in respect of deleted cell frequency in blood lymphocytes.
| Parameters | <1/3 deleted cells | >1/3 to <2/3 deleted cells | >2/3 deleted cells | Remarks |
|---|
| Number of cases | 4 | 5 | 2 | |
| Cono-truncal cardiac anomaly such as Fallot’s tetralogy (TOF), interrupted aortic arch, truncus arteriosus or major aorto-pulmonary collateral arteries / suspected but not evaluated due to intensive care | 3/4*(TOF) | 5/5(TOF in 3) | 2/2(TOF) | Cardiac abnormality does not correlate on concentration of deleted cells in blood lymphocytes |
| Dysmorphism (broad bulbous nose, square shaped tip of nose, short philtrum, telecanthus, slanting eyes, low set ears, etc.,) | 3/4* | 5/5 | 2/2 | Dysmorphism also does not correlate on concentration of deleted cells in blood lymphocytes |
| Hypocalcaemia/seizures/etc., | 2/4 | 1/5 | 0/2 | Hypocalcaemia also does not correlate on concentration of deleted cells in blood lymphocytes |
| Immunodeficiency or thymic hypoplasia | 1/4(hypoplastic thymus) | 0/5 | 0/2 | Thymic hypoplasia also does not correlate on concentration of deleted cells in blood lymphocytes |
| Cleft palate, high arched palate, velopharyngeal insufficiency or swallowing difficulty | 3/4* | 2/5 | 2/2 | Cleft palate, high arched palate, velopharyngeal insufficiency or swallowing difficulty also does not correlate on concentration of deleted cells in blood lymphocytes |
| Others (long slender fingers/hands, short stature, renal abnormalities, etc.,) | 3/4* | 5/5 | 2/2 | Prevalence little more with >35% deletions in blood lymphocytes |
| Family history of congenital cardiac defects | 2/4 | 0/5 | 0/2 | Family history of congenital cardiac defects observed only in cases with <1/3 deleted cell line |
| Remarks | | | | Clinical presentation does not correlate with deletion frequency in lymphocytes |
* One baby of 1½ months old was very sick with central cyanosis in ICU thus not evaluate for dysmorphism, pharyngeal insufficiency or cardiac malformation although major cardiac malformation was suspected clinically (also F/H of cardiac defect)
Our experience with SNP microarray (outsourced) was unsatisfactory for mosaicism detection in cases with low grade mosaicism. However, others have demonstrated that microarray is capable of detecting low level of mosaicism, as low as 25% [44-46] or levels as low as 10% but only under optimal conditions [47]. Some additional studies suggest that BAC based arrays CGH are more reliable to detect low level of mosaicism (at level of 10%) than oligo array (at level of 20%) [44]. However, for prenatal diagnosis of microdeletion mosaicism requires about 30% concentration of deleted cell line on BoBs (BAC on beads) array [48]. SNP array is more sensitive and can detect chromosomal mosaicism at levels as low as 5% [49] but for reliable detection of mosaicism higher concentration is required, preferably >25% [50]. Similarly, sensitivity of detecting mosaicism by QF-PCR/MLPA is lower than microarray although both methods can detect mosaic aneuploidy as low as 10% [48]. Our experimentation with QF PCR on mosaic deletion (<50%) cases was unreliable due to wide variation in amplification (allele to allele, individual to individual). Conventional cytogenetic analysis of metaphase cells can also provide information about mosaicism but may not accurately reflect levels and distribution of mosaicism (culture biasness and low number of metaphase cell count). FISH analysis of uncultured sample provides most reliable results to study mosaicism as this overcome culture artifact/clonal selection besides ability to analyse large numbers of cells of various types and origin rapidly thereby interphase FISH serves as the best method to detect mosaicism, even at low-grade.
Conclusion
We conclude that the high prevalence of mosaicism (28.2%) in 22q11.2 microdeletion (DiGeorge) syndrome, often low-grade (36% cases) and frequently associated with somatic mosaicism. Interphase FISH analysis is the best available method to detect mosaicism, particularly for low grade. Phenotypic variability in microdeletion syndrome is complex and does not depend solely on mosaicism.
Ethics approval and consent to participate: This study has been approved by the institutional ethics committee of All India Institute of Medical Sciences. Procedure was in accordance with the Helsinki declaration. Informed consent was obtained from the patient in the accordance with the requirement of the institutional ethics committee.
TOF: Tetralogy of fallot; Dys: Dysmorphism; DD: Developmental delay; HAP: High arched palate; LSF: Long slender fingers; CP: Cleft palate; NPI: Nasopharyngeal insufficiency; MCK: Multicystic kidney; CHD: Congenital heart defect; VSD: Ventricular septal defect; PDA: Patent ductus arteriosus; HC: Hypocalcemia; F/H: Family history; ICU: Intensive care unit; TH: Thymic hypoplasia; CL: Cleft lip; SD: Skeletal deformity
* One baby of 1½ months old was very sick with central cyanosis in ICU thus not evaluate for dysmorphism, pharyngeal insufficiency or cardiac malformation although major cardiac malformation was suspected clinically (also F/H of cardiac defect)
[1]. Oskarsdottir S, Vujic M, Fasth A, Incidence and prevalence of the 22q11 deletion syndrome: A population-based study in Western SwedenArch Dis Child 2004 89(2):148-51.10.1136/adc.2003.02688014736631 [Google Scholar] [CrossRef] [PubMed]
[2]. Franke UC, Scambler PJ, Loffler C, Lons P, Hanefeld F, Zoll B, Interstitial deletion of 22q11 in DiGeorge syndrome detected by high resolution and molecular analysisClin Genet 1994 46(2):187-92.10.1111/j.1399-0004.1994.tb04222.x7820929 [Google Scholar] [CrossRef] [PubMed]
[3]. Halder A, Fauzdar A, Kabra M, Saxena A, Detection of 22q11.2 hemizygous deletion by interphase FISH in a patient with features of CATCH22 SyndromeInd Pediatr 2005 42(12):1236-39. [Google Scholar]
[4]. Halder A, Jain M, Chaudhary I, Kabra M, Prevalence of 22q11.2 microdeletion in 146 patients with cardiac malformation in a referral hospital of North IndiaBMC Medical Genetics 2010 11:10110.1186/1471-2350-11-10120573211 [Google Scholar] [CrossRef] [PubMed]
[5]. Halder A, Jain M, Chaudhary I, Gupta N, Kabra M, Fluorescence in-situ hybridization (FISH) using non-commercial probes in the diagnosis of clinically suspected microdeletion syndromes: an experience with 301 casesInd J Med Res 2013 138:135-42. [Google Scholar]
[6]. Mantripragada KK, Tapia-Paez I, Blennow E, Nilsson P, Wedell A, Dumanski JP, DNA copy-number analysis of the 22q11 deletion-syndrome region using array-CGH with genomic and PCR-based targetsInt J Mol Med 2004 13(2):273-79.10.3892/ijmm.13.2.27314719134 [Google Scholar] [CrossRef] [PubMed]
[7]. Halder A, Jain M, Chaudhary I, Varma B, Chromosome 22q11.2 microdeletion in monozygotic twins with discordant phenotype and deletion sizeMolecular Cytogenetics 2012 5(1):1310.1186/1755-8166-5-1322413934 [Google Scholar] [CrossRef] [PubMed]
[8]. Halder A, Jain M, Kalsi AP, SNP microarray in FISH negative clinically suspected 22q11.2 microdeletion syndromeScientifica (Cairo) 2016 2016:582643110.1155/2016/582643127051557 [Google Scholar] [CrossRef] [PubMed]
[9]. Jianrong L, Yinglong L, Xiaodong L, Cuntao Y, Bin C, Bo W, 22q11.2 deletion mosaicism in patients with conotruncal heart defectsBirth Defects Res a Clin Mol Teratol 2006 76(4):262-65.10.1002/bdra.2024616575883 [Google Scholar] [CrossRef] [PubMed]
[10]. Jalali GR, Vorstman JA, Errami A, Vijzelaar R, Biegel J, Shaikh T, Detailed analysis of 22q11.2 with a high density MLPA probe setHum Mutat 2008 29(3):433-40.10.1002/humu.2064018033723 [Google Scholar] [CrossRef] [PubMed]
[11]. Gao M, Pang H, Zhao Y, Li-Ling J, Analysis of genomic copy number variations in 36 fetuses with heart malformations using next-generation sequencingZhonghua Yi Xue Yi Chuan Xue Za Zhi 2017 34(4):524-27. [Google Scholar]
[12]. Russo CD, Di Giacomo G, Cignini P, Padula F, Mangiafico L, Mesoraca A, Comparative study of aCGH and Next Generation Sequencing (NGS) for chromosomal microdeletion and microduplication screeningJ Prenat Med 2014 8(3-4):57-69.10.4172/2165-7920.1000455 [Google Scholar] [CrossRef]
[13]. Dempsey MA, Schwartz S, Waggoner DJ, Mosaicism del (22) (q11.2q11.2)/ dup (22) (q11.2q11.2) in a patient with features of 22q11.2 deletion syndromeAm J Med Genet A 2007 143A(10):1082-86.10.1002/ajmg.a.3169717431914 [Google Scholar] [CrossRef] [PubMed]
[14]. Halder A, Jain M, Kabra M, Gupta N, Mosaic 22q11.2 microdeletion syndrome: diagnosis and clinical manifestations of two casesMolecular Cytogenetics 2008 1:1810.1186/1755-8166-1-1818691436 [Google Scholar] [CrossRef] [PubMed]
[15]. Prabhu S, Jenny B, James H, Provenzano S, Mosaic 22q11.2 deletion and tetralogy of Fallot with absent pulmonary valve: an unreported associationWorld J Pediatr Congenit Heart Surg 2015 6(2):342-45.10.1177/215013511456168625870364 [Google Scholar] [CrossRef] [PubMed]
[16]. Chaddha V, Agarwal S, Phadke SR, Halder A, Low level of mosaicism in atypical Prader Willi syndrome: detection using fluorescent in situ hybridizationIndian Pediatr 2003 40(2):166-68. [Google Scholar]
[17]. Chen CP, Chern SR, Lee CC, Lin SP, Chang TY, Wang W, Prenatal diagnosis of mosaic 22q11.2 microdeletionPrenat Diagn 2004 24(8):660-62.10.1002/pd.91715305360 [Google Scholar] [CrossRef] [PubMed]
[18]. Consevage MW, Seip JR, Belchis DA, Davis AT, Baylen BG, Rogan PK, Association of a mosaic chromosomal 22q11 deletion with hypoplastic left heart syndromeAm J Cardiol 1996 77(11):1023-25.10.1016/S0002-9149(97)89165-5 [Google Scholar] [CrossRef]
[19]. Patel ZM, Gawde HM, Khatkhatay MI, 22q11 microdeletion studies in the heart tissue of an abortus involving a familial form of congenital heart diseaseJ Clin Lab Anal 2006 20(4):160-63.10.1002/jcla.2012516874809 [Google Scholar] [CrossRef] [PubMed]
[20]. Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental Neurofibromatosis Type-1 (NF1)Hum Mutat 2011 32(2):213-19.10.1002/humu.2141821280148 [Google Scholar] [CrossRef] [PubMed]
[21]. Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 geneAm J Hum Genet 2004 75(3):410-23.10.1086/42362415257518 [Google Scholar] [CrossRef] [PubMed]
[22]. Koolen DA, Dupont J, de Leeuw N, Vissers LE, van den Heuvel SP, Bradbury A, Two families with sibling recurrence of the 17q21.31 microdeletion syndrome due to low-grade mosaicismEur J Hum Genet 2012 20(7):729-33.10.1038/ejhg.2012.122293690 [Google Scholar] [CrossRef] [PubMed]
[23]. Petek E, Jenne DE, Smolle J, Binder B, Lasinger W, Windpassinger C, Mitotic recombination mediated by the JJAZF1 (KIAA0160) gene causing somatic mosaicism and a new type of constitutional NF1 microdeletion in two children of a mosaic female with only few manifestationsJ Med Genet 2003 40(7):520-25.10.1136/jmg.40.7.52012843325 [Google Scholar] [CrossRef] [PubMed]
[24]. Willemsen MH, Beunders G, Callaghan M, de Leeuw N, Nillesen WM, Yntema HG, Familial Kleefstra syndrome due to maternal somatic mosaicism for interstitial 9q34.3 microdeletionsClin Genet 2011 80(1):31-38.10.1111/j.1399-0004.2010.01607.x21204793 [Google Scholar] [CrossRef] [PubMed]
[25]. Goh ESY, Banwell B, Stavropoulos DJ, Shago M, Yoon G, Mosaic microdeletion of 17p11.2–p12 and duplication of 17q22–q24 in a girl with Smith–Magenis phenotype and peripheral neuropathyAm J Med Genet Part A 2014 164A(3):748-52.10.1002/ajmg.a.3632224357149 [Google Scholar] [CrossRef] [PubMed]
[26]. Petrin AL, Daack-Hirsch S, L’Heureux J, Murray JC, A case of 3q29 microdeletion syndrome involving oral cleft inherited from a nonaffected mosaic parent: molecular analysis and ethical implicationsCleft Palate Craniofac J 2011 48(2):222-30.10.1597/09-14920500065 [Google Scholar] [CrossRef] [PubMed]
[27]. Al-Zahrani J, Al-Dosari N, Abudheim N, Alshidi TA, Colak D, Al-Habit O, Chromosome 12q24.31-q24.33 deletion causes multiple dysmorphic features and developmental delay: First mosaic patient and overview of the phenotype related to 12q24qter defectsMol Cytogenet 2011 4:910.1186/1755-8166-4-921457577 [Google Scholar] [CrossRef] [PubMed]
[28]. Tekin M, Jackson-Cook C, Buller A, Ferreira-Gonzalez A, Pandya A, Garrett CT, Fluorescence in situ hybridization detectable mosaicism for Angelman syndrome with biparental methylationAm J Med Genet 2000 95(2):145-49.10.1002/1096-8628(20001113)95:2<145::AID-AJMG10>3.0.CO;2-R [Google Scholar] [CrossRef]
[29]. Anderlid BM, Lundin J, Malmgren H, Lehtihet M, Nordgren A, Small mosaic deletion encompassing the snoRNAs and SNURF-SNRPN results in an atypical Prader-Willi syndrome phenotypeAm J Med Genet A 2014 164A(2):425-31.10.1002/ajmg.a.3630724311433 [Google Scholar] [CrossRef] [PubMed]
[30]. Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probesAm J Hum Genet 2000 66(1):100-09.10.1086/30270910631140 [Google Scholar] [CrossRef] [PubMed]
[31]. Laufer-Cahana A, Krantz ID, Bason LD, Lu FM, Piccoli DA, Spinner NB, Alagille syndrome inherited from a phenotypically normal mother with a mosaic 20p microdeletionAm J Med Genet 2002 112(2):190-93.10.1002/ajmg.1061612244554 [Google Scholar] [CrossRef] [PubMed]
[32]. Eckmann-Scholz C, Gesk S, Nagel I, Haake A, Bens S, Heidemann S, Conflicting results of prenatal FISH with different probes for Down’s Syndrome critical regions associated with mosaicism for a de novo del (21) (q22) characterised by molecular karyotyping: Case reportMol Cytogenet 2010 3:1610.1186/1755-8166-3-1620815924 [Google Scholar] [CrossRef] [PubMed]
[33]. Kasprzak L, Der Kaloustian VM, Elliott AM, Shevell M, Lejtenyi C, Eydoux P, Deletion of 22q11 in two brothers with different phenotypeAm J Med Genet 1998 75(3):288-91.10.1002/(SICI)1096-8628(19980123)75:3<288::AID-AJMG12>3.0.CO;2-L [Google Scholar] [CrossRef]
[34]. Blennow E, Lagerstedt K, Malmgren H, Sahlén S, Schoumans J, Anderlid B, Concurrent microdeletion and duplication of 22q11.2Clin Genet 2008 74(1):61-67.10.1111/j.1399-0004.2008.01008.x18445048 [Google Scholar] [CrossRef] [PubMed]
[35]. Phelan MC, Deletion 22q13.3 syndromeOrphanet J Rare Dis 2008 3:1410.1186/1750-1172-3-1418505557 [Google Scholar] [CrossRef] [PubMed]
[36]. Tobias ES, Morrison N, Whiteford ML, Tolmie JL, Towards earlier diagnosis of 22q11deletionsArch Dis Child 1999 81(6):513-14.10.1136/adc.81.6.51310569971 [Google Scholar] [CrossRef] [PubMed]
[37]. Mascarello JT, Hirsch B, Kearney HM, Ketterling RP, Olson SB, Quigley DI, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. Section E9 of the American College of Medical Genetics technical standards and guidelines: fluorescence in situ hybridizationGenet Med 2011 13(7):667-75.10.1097/GIM.0b013e318222729521738013 [Google Scholar] [CrossRef] [PubMed]
[38]. Halder A, Fauzdar A, Potential use of blood, buccal and urine cells for rapid noninvasive diagnosis of suspected aneuploidy using FISHJournal of Clinical & Diagnostic Research 2007 1(2):32-38. [Google Scholar]
[39]. McDonald-McGinn DM, Zackai EH, Genetic counseling for 22q11.2 deletion developmental disabilitiesResearch Reviews 2008 14(1):69-74.10.1002/ddrr.1018636638 [Google Scholar] [CrossRef] [PubMed]
[40]. Piotrowski A, Bruder CE, Andersson R, Diaz de Stahl T, Menzel U, Sandgren J, Somatic mosaicism for copy number variation in differentiated human tissuesHum Mutat 2008 29(9):1118-24.10.1002/humu.2081518570184 [Google Scholar] [CrossRef] [PubMed]
[41]. Kaplan L, Foster R, Shen Y, Parry DM, McMaster ML, O’Leary MC, Monozygotic twins discordant for neurofibromatosis 1Am J Med Genet A 2010 152A(3):601-06.10.1002/ajmg.a.3327120186797 [Google Scholar] [CrossRef] [PubMed]
[42]. Cassidy SB, Thuline HC, Holm VA, Deletion of chromosome 15 (q11-13) in a Prader-Labhart-Willi syndrome clinic populationAm J Med Genet 1984 17(2):485-95.10.1002/ajmg.13201702116336316 [Google Scholar] [CrossRef] [PubMed]
[43]. Malzac P, Moncla A, Pedeillier K, Vo Van C, Girardot L, Voelckel MA, Atypical molecular findings identify limits of technical screening tests for Prader-Willi and Angelman syndrome diagnosesAm J Med Genet 1998 78(3):242-44.10.1002/(SICI)1096-8628(19980707)78:3<242::AID-AJMG6>3.0.CO;2-R [Google Scholar] [CrossRef]
[44]. Ballif BC, Rorem EA, Sundin K, Lincicum M, Gaskin S, Coppinger J, Detection of low-level mosaicism by array CGH in routine diagnostic specimensAm J Med Genet A 2006 140(24):2757-67.10.1002/ajmg.a.3153917103431 [Google Scholar] [CrossRef] [PubMed]
[45]. Novik V, Moulton EB, Sisson ME, Shrestha SL, Tran KD, Stern HJ, The accuracy of chromosomal microarray testing for identification of embryonic mosaicism in human blastocystsMol Cytogenet 2014 7(1):1810.1186/1755-8166-7-1824581286 [Google Scholar] [CrossRef] [PubMed]
[46]. Valli R, Marletta C, Pressato B, Montalbano G, Lo Curto F, Pasquali F, Comparative genomic hybridization on microarray (a-CGH) in constitutional and acquired mosaicism may detect as low as 8% abnormal cellsMol Cytogenet 2011 4:1310.1186/1755-8166-4-1321554683 [Google Scholar] [CrossRef] [PubMed]
[47]. Cohen AS, Wilson SL, Trinh J, Ye XC, Detecting somatic mosaicism: considerations and clinical implicationsClin Genet 2015 87(6):554-62.10.1111/cge.1250225223253 [Google Scholar] [CrossRef] [PubMed]
[48]. Cheng YK, Wong C, Wong HK, Leung KO, Kwok YK, Suen A, The detection of mosaicism by prenatal BoBs™Prenat Diagn 2013 33(1):42-49.10.1002/pd.400623168997 [Google Scholar] [CrossRef] [PubMed]
[49]. Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysisHum Mol Genet 2010 19(7):1263-75.10.1093/hmg/ddq00320053666 [Google Scholar] [CrossRef] [PubMed]
[50]. Northrop LE, Treff NR, Levy B, Scott RT Jr, SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocystsMol Hum Reprod 2010 16(8):590-600.10.1093/molehr/gaq03720479065 [Google Scholar] [CrossRef] [PubMed]