Granular cell tumours are rare neoplasms that occur in almost all parts of the body, ranging from dermis, subcutaneous tissue, breast and even the gastrointestinal tract [1]. Their schwannian origin has been well established, by ancillary techniques including IHC and electron microscopy [2]. GCT was first described by Abrikossoff in 1926 as myoblastoma (myoblastenmyome) due to the probable origin from skeletal muscles [1,3,4]. It is more often benign, involving the skin, subcutaneous tissue and mucosal surfaces, although malignant change has been described [3,4], the latter diagnosis being based on criteria established by Fanburg-Smith JC et al., [5]. The six criteria established by Fanburg-Smith JC et al., include the presence of necrosis, nuclear pleomorphism, high nucleo-cytoplasmic ratio, vesicular nuclei with large nucleoli, spindling of tumour cells and increased mitotic activity (>2 mitoses per 10 HPF at 200X magnification) [5]. The authors hereby describe a series of 22 cases of GCT, diagnosed over a period of five years. Since, there is no similar study conducted, the authors wanted to check for demographics of the present rare condition. The clinical features, histology and diagnostic difficulties have been described in detail.
Materials and Methods
The present retrospective study conducted in the Department of Pathology, Kasturba Medical College, Manipal, Karnataka, India, a tertiary care center in Southern India, from 2012 to 2017. After obtaining clearance by Institutional Ethical Committee (IEC-346/18), 23 cases of GCT were retrieved from the digital records, archived in the department. All available slides were retrieved, reviewed and a case reported as GCT in bladder was reclassified as bladder paraganglioma and excluded from the series. Hence, a total of 22 cases were included with a histopathological diagnosis consistent with GCT. In addition to Haematoxylin and Eosin (H&E) sections, Periodic Acid Schiff (PAS) and reticulin stains were also studied. In a few cases, IHC slides were also available for evaluation.
Results
The average age at presentation of GCTs in the current series was 33.6 years, ranging from six months to 74 years. Women were more often affected than males with a female to male ratio of 3.4:1 (17 to 5). Most common organ involved was skin and subcutaneous tissue (14), followed by lip and tongue (4) and breast (2), one case each was documented in oesophagus and vulva.
Clinical diagnosis ranged from lipoma, neurofibroma to sebaceous cyst in case of dermal or subcutaneous lesions. Both the cases in the breast were suspected to be malignant breast tumours. Lesional size ranged from less than a centimetre dimension (0.5×0.5 cm) to 2×1.5 cm. On follow-up, varying from a period of two to six months, all the patients were symptom-free, without any recurrences. The results are tabulated in [Table/Fig-1].
Clinicopathological features of the cases.
| S No. | Age (years) | Sex | Site | Clinical diagnosis | Size | IHC |
|---|
| 1 | 1 | Female | Skin | - | 0.5×0.5 cm | |
| 2 | 11 | Female | Skin | - | 0.8×0.8 cm | S-100 + |
| 3 | 42 | Female | Skin-suprapubic area | - | 1×0.5 cm | |
| 4 | 27 | Male | Oesophagus | Malignancy | 0.5×0.5 cm | |
| 5 | 28 | Female | Breast | Malignancy | 4×4 cm | CD68 & S100 + |
| 6 | 50 | Female | Left foot | Infected callus | 1.8×2 cm | |
| 7 | 21 | Male | Left forearm-SC | - | 0.5×0.5 cm | |
| 8 | 41 | Female | Muscle lump | - | 0.5×0.5 cm | |
| 9 | 33 | Female | Tongue | - | 0.5×0.5 cm | |
| 10 | 31 | Female | Subcutaneous tissue | Neurofibroma | 0.5×0.5 cm | |
| 11 | 28 | Female | Tongue | leiomyoma | 1×0.8 cm | |
| 12 | 33 | Female | Soft tissue | Lipoma | 2.5×2.5 cm | |
| 13 | 50 | Female | Middle finger | - | 1×1 cm | |
| 14 | 10 | Male | Lower back | Sebaceous cyst | 0.5×0.5 cm | |
| 15 | 34 | Female | Flank | Lipoma | 1×0.5 cm | |
| 16 | 44 | Female | Supraclavicular region | Sebaceous cyst | 2×1.5×0.5 cm | |
| 17 | 41 | Female | Breast | Malignancy | 1.3×1.2 cm | CD68 & S100 + |
| 18 | 36 | Female | Lateral aspect of lip | Mucous cyst | 0.6×0.5 cm | |
| 19 | 18 | Male | Skin punch biopsy | Neurofibroma | 3.5 mm across | S100 + |
| 20 | 74 | Male | Neck | Lymph node | 1.5×1 cm | |
| 21 | 68 | Female | Labium Majus | Ca Vulva | 2×1.5 cm | |
| 22 | 18 | Female | Lateral border of tongue | Fibroma | 1×0.8 cm | |
cm=centimeter
Microscopy
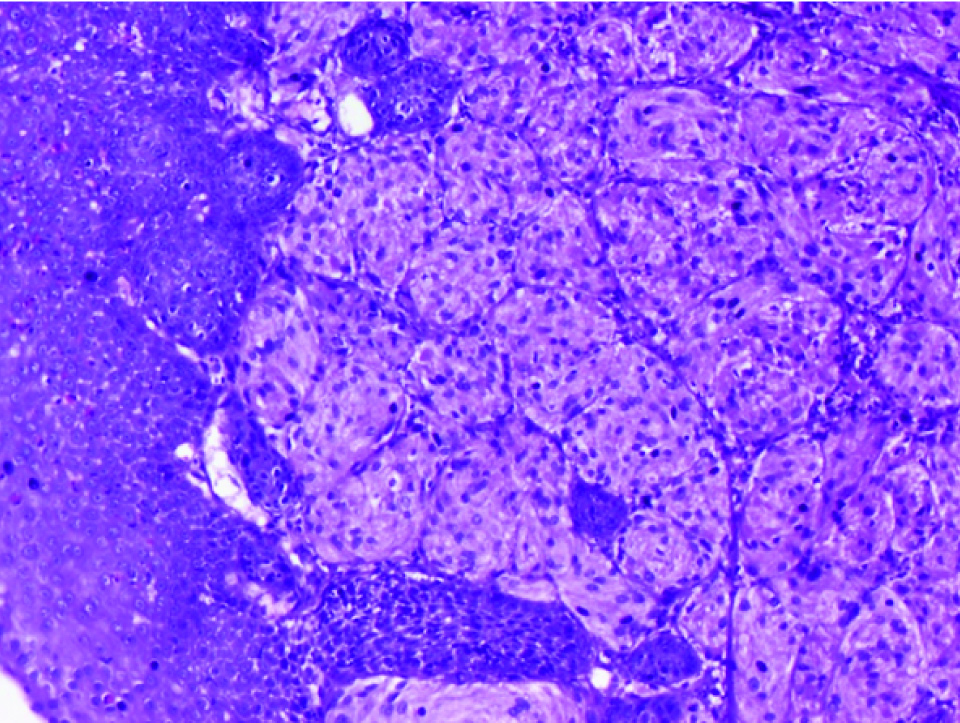
All the lesions, except one, were ill-circumscribed with infiltrative margins. Lesions involving the dermis sometimes, extended up to the subcutaneous fat [Table/Fig-2]. The tumour cells were characterised by moderate to abundant pale eosinophilic finely granular cytoplasm, round nuclei and pustule-ovoid bodies of Milian [Table/Fig-3]. Mitoses were very rare {1 mitoses/ 10 high power field (hpf)}. Overlying pseudoepitheliomatous hyperplasia was noted in 11 cases (50%). Entrapped adnexae, nerves and lymphoid aggregates were other associated findings [Table/Fig-4]. One lesion showed the presence of giant cells. Lesion involving the lip showed prominent interspersed capillaries, while in one case, perivascular infiltration by tumour cells was noted [Table/Fig-5]. None of the case showed necrosis or other features of malignancy as described by Fanburg-Smith JC et al., [5]. The microscopic findings have been tabulated in [Table/Fig-6].
Stratified squamous epithelium with a tumour composed of nests of granular cells (Haematoxylin and eosin, 10X).
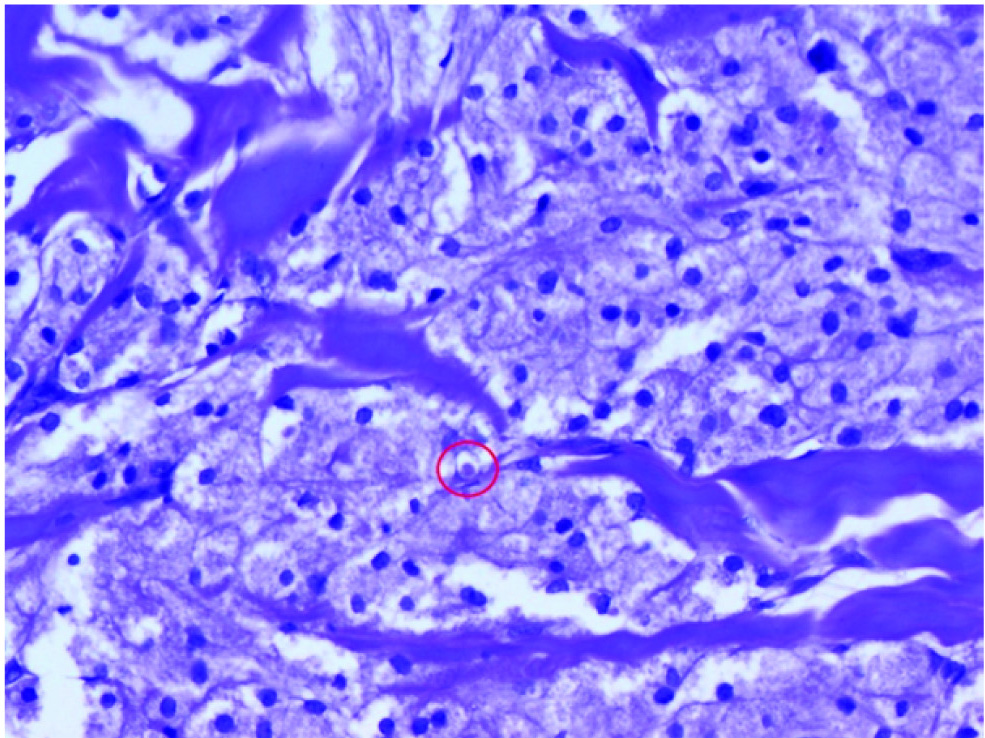
Cells with abundant granular cytoplasm, central oval nucleus, with pustule-ovoid bodies of Milian (in a red circle) (Haematoxylin and eosin, 20X).
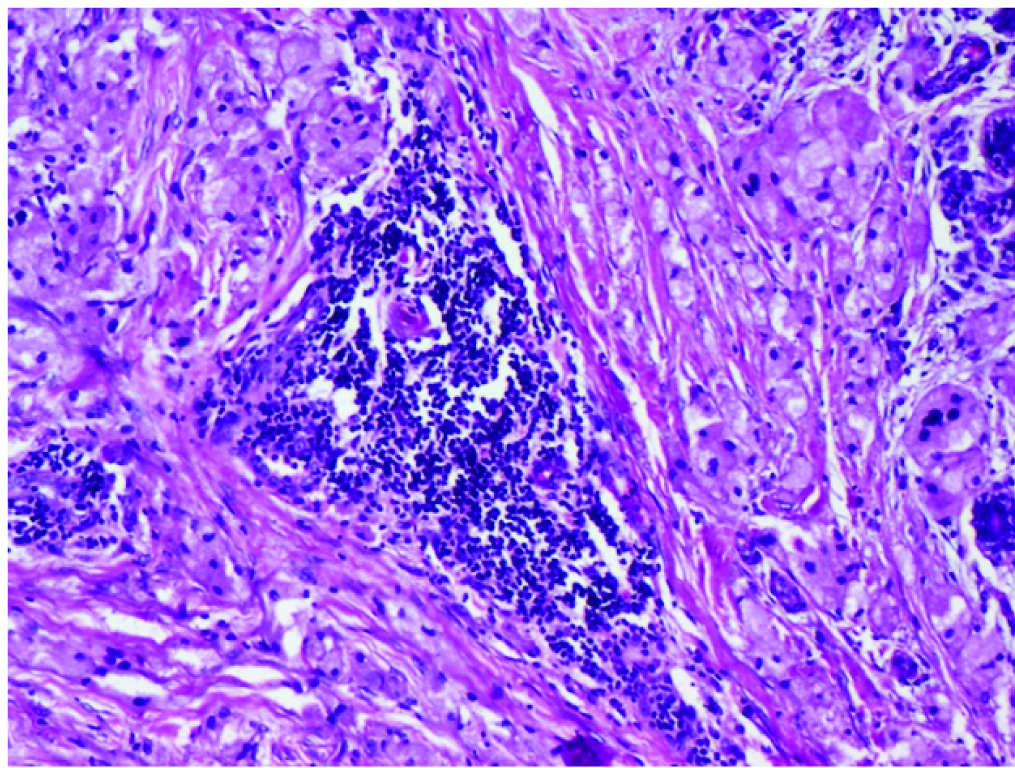
Granular cells with an aggregate of tumour infiltrating lymphocytes (Haematoxylin and eosin, 10X).
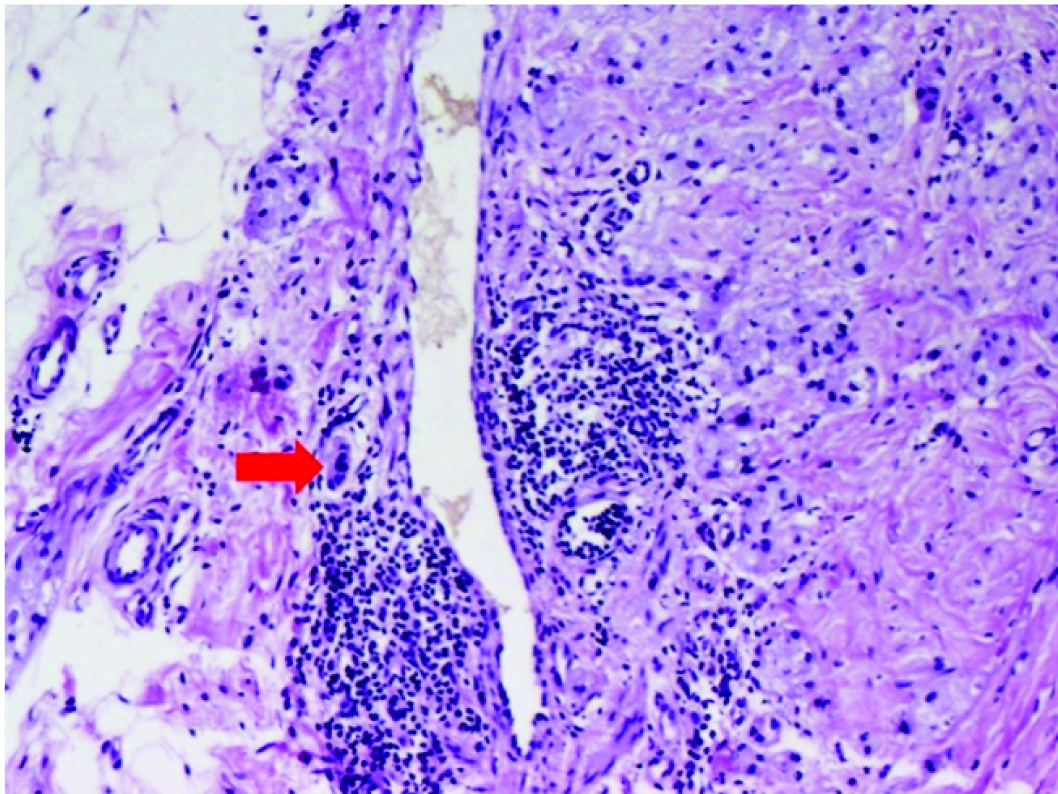
Invasion of the vessel wall along with lymphocytic infiltration (red arrow) (Haematoxylin and eosin, 10X)
| S No. | Infiltrative margin | PEH | Intranuclear inclusions | Lymphocytic aggregate | Perineural (PN)/ intra/perivascular (PV) invasion | Entrapped nerve/adipocytes/adnexae | Any other feature |
|---|
| 1 | + | - | + | ++ | + PN | - | - |
| 2 | + nodular pattern | + | - | ++ | + PN | + | Nodules + |
| 3 | + | + area overlying tumour cells | - | - | - | - | Anisonucleosis + |
| 4 | + | + | - | + | - | - | Whorling + |
| 5 | + | - | - | - | - | - | - |
| 6 | + | + | - | - | - | + | Alveolar pattern |
| 7 | + | - | - | - | - | - | |
| 8 | + | - | - | ++ | + PN | - | Nodules + |
| 9 | + (skeletal muscle) | + | - | - | - | Peripheral nerves + | - |
| 10 | + | - | - | ++ | + PN | + (both) | |
| 11 | + | + | - | - | - | Peripheral nerves+ | Very few clusters of tumour cells |
| 12 | + | - | + | ++ | +(PV) | + | Pacinian corpuscle+, nodules |
| 13 | + | - (epithelium +) | - | + | - | - | Giant cells (tumour) +; bizarre nuclei + |
| 14 | + | - | + | + | - | Adnexae + | Nodules + |
| 15 | Circumscribed | - | Prominent nucleoli | - | - | - | Occasional bizarre nuclei |
| 16 | + | -(epithelium+) | + | + | + PV | + | Adjacent nerves |
| 17 | + | - | + | ++ | - | + | - |
| 18 | + | + | - | - | - | Peripheral nerves+ | Small tumour with nodules of 5-6 cells each |
| 19 | + | + | - | - | + PV | + | - |
| 20 | + | + | - | - | - | - | - |
| 21 | + | + | - | - | - | - | Bizarre cells, ulceration + |
| 22 | + | + | - | - | - | - | Lobular pattern |
+ present; - absent; ++ more than one in number
Immunohistochemistry
Immunohistochemistry studies were performed in four of the 22 cases (i.e., case number 2, 5, 18 and 20). Tumour cells were positive for S-100 protein and CD68, two cases each in breast and skin.
Discussion
The present retrospective study of the clinicopathological features of GCT, range of sites affected, morphological variations, differential diagnosis based on site and to evaluate if associated findings like Pseudoepitheliomatous Hyperplasia (PEH) will be helpful in diagnosis of the entity.
Patient’s age at presentation of GCT is between 32 to 38 years [6]. These are known with a female preponderance (1.8-2.9:1) [6]. The findings are similar in the current series. Skin and subcutaneous tissue was the most common site (63.6%), followed by tongue (63.6%) and breast (9.1%). Present findings are very similar to the observations by Lack EE et al., [1]. Oesophagus, which has been described as a common site in literature, was uncommon in the present series [1]. A rare site of GCT which has been described in literature is the labium majus [7]. We came across, one such case in the current series as well.
Though, histologic diagnosis of GCT is often straight forward, it is rarely suspected clinically. The variation in histologic differentials is site dependent. For example, in the salivary glands, oncocytic neoplasms of salivary gland origin may be the primary differential diagnosis considered, while in the breast parenchyma, they may be confused with apocrine carcinomas. However, in the spermatic cord, ectopic Leydig cells are the main differential diagnostic consideration [5]. Some authors opine that the round to ovoid eosinophilic cytoplasmic granules surrounded by clear halo, called the pustule-ovoid bodies of Milian are quite specific and can be used to exclude other differentials [8]. We found pustule-ovoid bodies in all 22 cases.
The optimal treatment of GCT is local surgical excision. A review of literature suggests absence of recurrence even when margins are positive [1,4]. Controversy exists regarding the nature of surgery. However, due to the occurrence of rare malignant GCT (accounting for 2% of cases), complete excision with margin clearance is advocated [9]. Lesions with two of the listed features are labelled atypical while three or more indicate malignancy of the six criteria described by Fanburg-Smith JC et al., for the diagnosis of malignancy [5]. However, in 2011, Nasser H et al., reviewed the same and proposed that two features were consistently accurate in predicting malignancy, namely necrosis and mitoses (>2 mitoses/10 high-power fields) [10]. However, these two criteria when used alone did not detect all the malignant cases, which had already metastasised to distant sites, as described in the study by Machado I et al., [8]. The authors did not observe the presence of these features together in present series. One case showed mitoses 1/10 HPFs. However, necrosis was absent. Over the years, like endocrine neoplasms, metastasis alone seems to be the confirmatory sign for malignancy. Therefore, Machado I et al., proposed the term “GCT with increased risk of metastasis” rather than malignant GCT to indicate tumours with unfavorable features histologically [8]. In the present series, the average follow-up period was six months and had no recurrences. No unfavourable histologic features were noted in the present series.
Battistella M et al., in a multicentre study found infiltration of arrector pili muscle and vascular invasion in 23% and perineural infiltration in 66% of their cases [11]. However, they found that none of these factors including vascular invasion (without any tumour embolus) were associated with recurrence or adverse prognosis. Invasion of the vessel wall was seen in three of the present cases while four other cases were associated with perineural infiltration [Table/Fig-6].
Tumours arising in the skin or mucosal surfaces are associated with PEH [1]. This feature was seen in 11 cases in present series, when tumour cells were in close proximity to the skin or mucosa. However, tumours away from the epidermis, i.e. involving the reticular dermis or subcutaneous fat did not show this feature. In other words, PEH is a good diagnostic clue in superficial lesions, it may not be helpful in deep seated GCTs. Further, PEH should not be misinterpreted as carcinoma especially in superficial biopsy. Another common feature is tumour infiltrating lymphocytes, probably suggesting immune response [6]. which was seen in 10 cases (45%), with some showing lymphoid aggregates as well.
Immunohistochemical markers useful in GCT are S100 protein and CD68 [1,5,12]. S100 positivity proves its origin from neural crest – peripheral nerve sheath. A variant called non-neural GCT has been described based on the absence of S100 [12]. CD68 positivity is attributed to the intracytoplasmic phagolysosomes and does not imply its histiocytic origin.
Limitation
Limitations of the present study include the small sample size, due to the rarity of the tumour and lack of longer period of follow-up. A study with wider panel of immunohistochemical markers including Ki67 index may be included in future studies.
Conclusion
Granular cell tumours is a group of neoplasms that can occur in various organs. Though most have a benign course, malignant GCT is known to occur. Lack of circumscription, infiltrative borders and invasion of the vessel wall are common and are not associated with increased malignant potential. Pustule-ovoid bodies of Milian and lymphoid aggregates are common histologic features.
cm=centimeter