Introduction
Microorganisms adapt to the environmental changes by modulating the molecular intra-cellular mechanisms and their gene(s) expression(s) parsimoniously. Further, when they are in persistent contact with the environment for longer duration, they can generate molecular genetic alterations which could enable them to adapt to the adverse environment. Under selective pressure, these variants can evolve, develop and dominate within a short duration. A good example for this molecular evolution is the emergence of drug resistance in the microorganisms during the prolonged antimicrobial therapy. The increasing pattern of multidrug resistance is a major morbidity and mortality threat in the management of both bacterial and/or fungal infectious diseases. High mortality and morbidity rates due to fungal pathogens are of great concern today, due to the limited therapeutic options. In the present review, we shall deal with fungal drug resistance, particularly Candida, as it is one of the most common fungus causing infections in humans with widespread prevalence and morbidity.
Candida species are the members of microbiota of human gastrointestinal and urinary tracts which could lead to diseases when the host immune mechanisms are compromised. Since, human’s immune system is able to counteract the candidal infections, the infections are usually asymptomatic. Candida albicans is a normal commensal of humans that resides in the oral cavity, gastrointestinal, vaginal and urinary tracts [1]. It acts as an opportunistic pathogen causing infections such as stomatitis, thrush, urinary tract-infections and can also cause severe systemic infections [1]. It causes infection when the host becomes debilitated or immunocompromised, the numbers of which are constantly increasing due to organ transplant, chemotherapy or due to the increase in prevalence of AIDS and hepatitis C [1,2]. Candida has various molecular mechanisms by which it maintains its character and pathogenicity. C. albicans has been found to be the predominant cause of invasive candidiasis [3]. However, in the recent years, many longitudinal studies have proved that Non Albicans species (NAC) have also been associated with clinical infections [4,5]. Candida is also isolated from nosocomial infections. Not only C.albicans, the other species of Candida like C. glabrata, C. tropicalis, C. krusei, C. parapsilosis are also found to cause hospital acquired infections [6,7]. As mentioned earlier, this could also be probably due to the rise in the number of immunocompromised patients, especially HIV cases, patients on immunosuppressive drugs and patients undergoing transplantation [3]. During the life long association with the human host, Candida generates genetically altered variants which could adapt to their new environment; the emergence of drug resistance is one of the major public health issues of global concern.
There are relatively few classes of antifungal drugs. This restricts clinician’s therapeutic usage and is further obliterated by the emergence of drug resistance [8]. The emergence of drug resistance in all pathogenic microorganisms, including fungi, is a process of evolution initiated by antimicrobial agents on prolonged exposure [9]. Whenever the pathogen population remains large even after drug treatment, the evolution of resistance becomes inevitable. The evolution of drug resistance depends on genetic variability, mostly mutation [9]. Resistance due to mutation arises in the pathogen by selection, genetic drift, recombination, and migration (including transmission between hosts). The emergence and spread of drug resistance depends on different possible mutations that enable the pathogen to avoid, remove or inactivate a drug [9]. The drug resistance can occur due to mutations in the drug targets, alteration in the sterol biosynthesis pathways, functional mutations in the transcription factors resulting in upregulation of ergosterol biosynthesis genes and multidrug efflux pumps. Genomic rearrangements resulting in gene amplification and loss of heterogenicity can also cause drug resistance.
Epidemiology of Candida infections: Centers for Diseases Control and Prevention (CDC) and the National Healthcare Safety Network has ranked Candida as fifth in causing hospital-acquired infections and fourth among Blood Stream Infection (BSI) pathogens [10]. Another international study by the SENTRY Antimicrobial Surveillance Program reported a total of 1239 candida BSI isolates from 79 medical centers in 2008-2009 [11]. This study reported high antifungal resistance among the C.glabrata isolates with the resistance rates to echinocandins (16.7%), fluconazole (16.7%), posaconazole (5.0%) and voriconazole (11.0%) in patients of 20-39 years age [11].
Candida species are approximately the fourth most common cause of nosocomial infections in ICUs, according to data from the National Nosocomial Infections Surveillance System and the European Prevalence of Infection in Intensive Care [12]. Candida may cause hospital acquired infection when its incidence is increased in hospitals or due to involvement of invasive interventions and long-term antibiotic usage [10]. The incidence of fungal infections varies widely with solid organ–transplant recipients; it ranges 5% in renal transplantation, 35% in lung and heart transplant recipients and up to 40% in liver transplantation [13]. Serious fungal infections affect nearly more than 1 billion people per year [14]. Cancer chemotherapy and allogeneic bone marrow transplantation may also be related to fungal disease, and 30% of acute leukaemia patients have invasive fungal infections [13].
However, community-acquired cases cannot be ignored. The SENTRY antimicrobial surveillance program detected 1,354 infection episodes related to Candida species between 2008 and 2009 among which community-acquired cases were 36.5% and they also reported community-acquired candidemia is higher in North America (63.5%) than in Europe (22.4%) [10]. The Asian picture, regarding incidence of candidemia is not very clear due to lack of multicentric studies [15]. A Southern India based study reported an incidence rate of 5.7% for candidemia among children with onco-haematological malignancies [16].
There is a lot of variation among the incidence and prevalence reports quoted from different parts of India. An incidence rate of 6.9% for Candida species in blood stream infections was reported by Sahni V et al., from Maulana Azad Medical College, New Delhi [17]. A study done in New Delhi gave a prevalence rate of 18% for Candida species among isolates of blood culture [18]. A similar 5-year study (2001-2005) by Xess I et al., from AIIMS, New Delhi, reported a prevalence rate of 6% for Candida species [19]. Another study from Rohtak, Northern India, reported an isolation rate of 8.1% for Candida species among neonatal septicaemia cases [20]. A 13-year study from a tertiary care hospital in Thailand showed a prevalence of 6.14% for Candida species among blood culture isolates [21]. Based on a study from SGPGI Lucknow, Candida species is stated as eighth among all isolates from BSI and reported an incidence rate of 1.61 per 1000 hospital admissions for candidemia [22].
Antifungal drugs in clinical treatment: Though, there are various classes of antifungal drugs, the classes of drug in current therapeutic use in Candida infections are relatively few [23,24]. Azoles, echinocandins, polyenes, nucleoside analogues and few other antifungal agents like allylamines, thiocarbamates are the various antifungal drugs in use. Azoles are the inhibitors of Lanosterol 14-α-Demethylase enzyme. The azole drugs include imidazoles (miconazole, econazole, clotrimazole, and ketoconazole) and triazoles (fluconazole, itraconazole, and voriconazole (second-generation, synthetic triazole derivative of fluconazole) and posaconazole (hydroxylated analogue of traconazole) [25,26]. Azoles are widely used for topical usage as well as in invasive Candidal infections [25]. Echinocandins are inhibitors of glycan synthesis. Caspofungin, micafungin, and anidulafungin are few echinocandins used in oesophageal and invasive candidiasis [27,28]. Polyenes act by binding to ergosterol. Polyenes like nystatin and amphotericin B have a broad spectrum antifungal action. Nucleoside analogues like flucytosine are inhibitors of DNA/RNA synthesis. Allylamines and thiocarbamates inhibit squaline epoxide enzyme used in the biosynthesis of ergosterol [29]. Griseofulvin is a tricyclic spirodiketone, acting by inhibiting fungal mitosis [Table/Fig-1].
Mechanism of action of antifungal drugs.
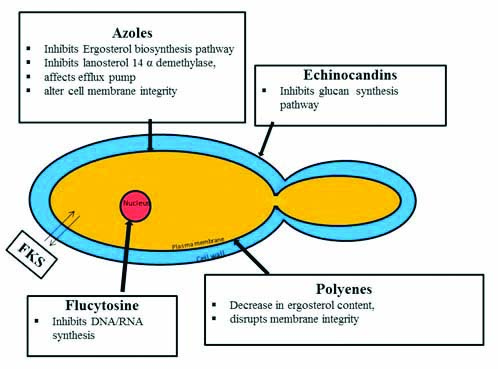
Antifungal Drug Resistance in Candida Species
Phenotype, genotype, serotype of the yeast, initial MICs, biofilm formation, fungistatic nature of the drug, drug dosage and pharmacokinetics, drug interactions, site and severity of the infections, immune status of the patient, noncompliance of the patient, etc., are amongst the various factors which may precipitate the emergence of drug resistance in the Candida species.
Resistance to azoles: Azoles are the largest family of antifungal drugs. Fluconazole, clotrimazole, ketoconazole, itraconazole, miconazole are some of the common antifungal drugs in azole group. Fluconazole is currently the widely used drug in treating the candidal infections. Resistance to these drugs is one of the major challenge in antifungal therapy and public health. Widespread, irrational and chronic usage of these drugs is one of the reasons for development of resistance to azole drugs.
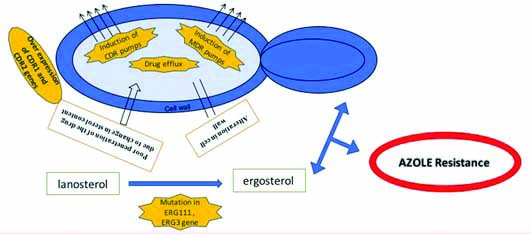
Azole group of drugs generally act by disrupting the fungal cell membrane by inhibiting an enzyme namely lanosterol 14-α-sterol Demethylase (DM) [26]. This enzyme is basically a cytochrome P 450 enzyme having heme cofactor azole binding site and is involved in the biosynthesis of ergosterol by mediating the conversion of lanosterol to ergosterol. Inhibition of this enzyme by azole leads to accumulation of the toxic product 14-α-methyl-3,6-diol and reduction in ergosterol content of the fungal plasma membrane which ultimately leads to disruption of the integrity of the fungal cell membrane resulting in reduced fungal growth. Various mechanisms of resistance to azoles have been reported in Candida species [30]. More than one type of resistance may be present in any strain which may even lead to cross resistance [31]. Any change in the target enzyme namely lanosterol 14-α-sterol demethylase may lead to development of drug resistance. Alterations in the affinity of azoles to the lanosterol 14-α-sterol demethylase enzyme may result in reduced binding of azoles to that enzyme [32,33]. Also, target site mutation or over expression of ERG11 genes (Ergosterol Biosynthesis enzyme-Lanosterol (C-14) demethylase) may lead to increased cellular content of 14 DM resulting in increased ergosterol synthesis. In addition, any alteration in the cell wall or the cell membrane may cause variation in the uptake of azoles. Any change in the sterol or phospholipid content of the plasma membrane may cause poor penetration of the drug into the membrane. Also, pumping out through over expressed efflux systems may cause decrease in the intracellular concentration of the azoles [32,33]. This induction of multidrug pumps resulting in decrease in concentration of drugs in the fungal cell membrane at the target enzyme lanosterol 14-α-sterol demethylase is one of the vital mechanisms of azole resistance.
Over expression of plasma membrane efflux pump is one of the major mechanisms of azole resistance. The major families of efflux proteins namely ABC and MFS are of great clinical significance in the azole resistance mechanisms. These proteins actively transport the compounds across the fungal cell membrane. ABC proteins are primary transporters and MFS are secondary transporters, both containing distinctive protein domains namely Nucleotide Binding Domains (NBDs) in ABC transporters and Transmembrane Domains (TMDs) in ABC and MFS transporters. Candida Drug Resistant (CDR) genes of the ATP-binding cassette superfamily and MDR genes of major facilitator super-family, encode the efflux pumps of the Candida species. Induction of CDR encoded efflux pumps has been found to be the common resistance mechanism in Candida to almost all azole groups of drugs, while induction of MDR encoded efflux pumps play an important role in fluconazole resistance [30].
Molecular studies have shown that the other important mechanisms of azole resistance in Candida is mutation of the genes encoding target enzyme ERG11 and over expression of the genes encoding for membrane transport proteins [32,34]. The ERG11 gene encodes for enzyme target lanosterol 14-α-sterol demethylase. Studies have shown several genetic alterations in ERG11 gene in Candida species. Any alteration or upregulation of this target enzyme may lead to azole resistance. Mutation in the ERG11 gene prevents binding of the azole drugs to the enzyme target [35]. It is said that the reduced affinity of ERG11 gene to fluconazole may be the cause for the intrinsic resistance exhibited by the C. krusei to the fluconazole [36]. However, it is reported that this mechanism plays a limited role in clinical resistance in Candida species to azoles [30]. Several point mutations have been identified in resistant strains of Candida species during the ERG11 gene sequence analysis. In a study, when the clinical strains of Candida were tested, azole resistance was found to be associated with a point mutation at amino acid 467 where arginine is replaced with lysine [37]. D116E and E266D are few other observed mutations, not necessarily associated with resistance [34]. However, few studies reported that though point mutation causing replacement of arginine and overexpression of ERG11 encoding the efflux pump systems are found in resistant strains, the overexpression of ERG11 gene may not be associated with azole resistance [34].
Another mechanism of azole resistance is development of bypass pathways. It is already mentioned that inhibition of lanosterol 14-α-sterol demethylase due to exposure to azoles may lead to the accumulation of the toxic product 14-α-methyl-3,6-diol. Mutations in the ERG3 gene prevents the formation of the toxic product 14-α-methyl-3,6-diol from 14-α-methylfecosterol [38]. Hence, if the ergosterol is replaced with the latter sterol, the fungal cell membrane will become functional and this may negate the azole leading to membrane disruptive effects.
Molecular studies revealed two types of efflux pumps namely ABC transporters and MFS proteins, responsible for the development of resistance to azoles in Candida species. The ABC transporter gene CDR and the MSF gene (CaMDR1 gene)-BEN-R are the genes for these transporters. In resistant strains, these genes are shown to be overexpressed. Few studies have reported that BEN-R gene is responsible for resistant to fluconazole in C.albicans [37,39]. CDR1 and CDR2 in C. albicans strains and CgCDR1 has been found to be responsible for resistance to azoles in C. glabrata [40]. Overexpression of CDR2 gene showing cross resistance to azoles in C. albicans have been reported [39,41] [Table/Fig-2].
Mechanism of resistance to azole group of drugs.
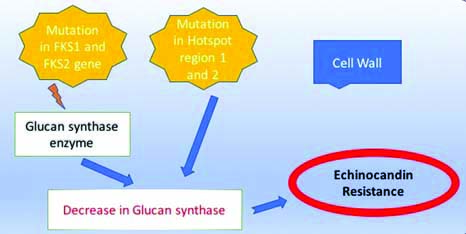
Resistance to echinocandins: β(1,3)D-glucan synthase is an enzyme which helps in the biosynthesis of β(1,3)D-glucan, an important component in the fungal cell wall [31]. Echinocandins act by inhibiting this β(1,3) D-glucan synthase enzyme causing defective fungal cell wall formation leading to cell death [31].
Point mutation and intrinsic mutation of the genes encoding FKS (Glucan synthase gene) subunits of the enzyme β(1,3)D-glucan synthase are responsible for the resistance or decrease in susceptibility to echinocandins [42]. There are some highly conserved regions in Candida species where the specific point mutation have been found to cluster around, known as “hot spot regions” [43]. Mutations in such hot spot regions may cause increase in Minimum inhibitory concentration (MIC), reduced β(1,3)D-glucan synthase sensitivity, and cross-resistance among the echinocandins [44,45].
In C.albicans, these hot spot regions namely HS1 is found in amino acid positions 641 to 649 and HS2 is found in amino acid positions 1345 to 1365 in FKS1 [44]. Such hot spot mutations FKS1 have also been reported in other species of Candida namely C. glabrata, C. dubliniensis, C. krusei, C. tropicalis [44,46,47]. Mutations in FKS2 (paralog of FKS1) can also lead to resistance to echinocandins in C. glabrata species [48]. However, C. parapsilosis, is less susceptible to echinocandins due to intrinsic mutation in FKS1. Studies have shown that C. parapsilosis exhibit a higher range of MIC values when compared with other Candida species [49].
Initiation of adaptive stress response is another mechanism of echinocandin resistance [50]. Studies report that when echinocandins inhibit β(1,3)D-glucan synthase enzyme, there is an increase in chitin synthesis in Candida species mediated by protein kinase C, high-osmolarity glycerol response and Ca2+ calcineurin signalling pathways [51,52]. Few studies have shown that few C. albicans strains can grow at supra-MIC concentrations of capsofungin by paradoxical effect [53-56]. These strains have high chitin levels at supra MIC concentration of capsofungin when compared to lower levels of capsofungin [54] [Table/Fig-3].
Mechanism of resistance to Echinocandins.
Resistance to polyenes: The commonly used polyenes in treating Candida infections are amphotericin B, nystatin, etc. Though there has been only a minimal resistance to amphotericin B documented in the literature over the past few decades; however, it is a matter of great concern. The major problem with the usage of this drug is the side effects and toxicity [57,58]. Resistance pattern differs with different species. Though, C. krusei and C. glabrata show a higher MIC to the polyenes than C. albicans, these species are often considered to be susceptible to amphotericin B.
Polyenes generally act by disrupting the fungal cytoplasmic membrane by interacting with ergosterol. Ergosterol is essential for maintaining the integrity of the fungal cell membrane and functioning of the membrane bound enzymes. The polyenes generate pores in the cell membrane through which potassium and magnesium ions of the cellular components escape and cause destruction of the proton gradient of the cell membrane finally leading to fungal cell death [57].
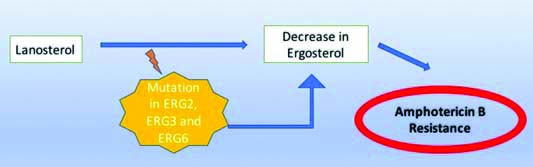
Polyenes have high affinity for ergosterol and low affinity for 3 hydroxy or oxosterols like fecosterol and episterol which is one of the important reasons in the emergence of resistance to polyenes drugs [59]. Studies reveal that polyenes favoured sterol namely ergosterol has been replaced with other biosynthetic precursors like lanosterol, episterol, fecosterol, lichesterol, etc. Though, this change may lead to overall increase in membrane sterol content but the availability of ergosterol to the polyenes may be affected significantly. This in turn may be one of the causes of development of resistance to the polyenes like amphotericin B.
Thus, the major causes of development of resistance to polyenes include inhibition of ergosterol synthesis leading to decrease in ergosterol levels, replacement of ergosterols with other biosynthetic precursors causing alteration in sterol content and change in the sterols phospholipids ratio [59,60]. Masking of the ergosterol in the cell membrane causing non-availability for binding with the polyenes is another mechanism of development of drug resistance [61]. In few resistant strains with no alteration in the membrane sterol content, the development of resistance is attributed to the change in the cell wall permeability to the polyenes. Increase in catalase activity of amphotericin B leading to decrease in the oxidative damage by the drug is also proposed to be another mechanism of drug resistance. Growth phase of the cell is also found to play a role in the development of resistance to polyenes. It is found that higher rate of breakdown and synthesis of cell wall occurs in the log phase leading to higher rate of access of amphotericin B to the cell membrane. However, there is lower rate of breakdown and synthesis of cell wall during the stationary phase of growth leading to the development of resistance to the polyenes.
It has been reported frequently that the polyene resistance is found to be higher in Candida non albicans species like C. tropicalis, C. glabrata, C. lusitaniae, C. parapsilosis, etc. C. glabrata is found to mutate frequently making it more likely to develop resistance to polyenes than the C. albicans [62].
Mutations in the genes encoding the enzymes involved in the synthesis of ergosterol may also lead to the development of resistance to the polyenes. C8 sterol isomerase and C5 sterol desaturase are the two important enzymes in the biosynthesis of ergosterol. C8 sterol isomerise is involved in the conversion of fecosterol into episterol. ERG2 gene regulate the activity of this enzyme. C5 sterol desaturase catalyses the episterol conversion into ergosterol. ERG3 genes encode this C5 sterol desaturase enzyme. Hence, any mutation or defect in the ERG2 and ERG3 gene may cause amphotericin B resistance. Clinical studies reported C.albicans resistant strains with ERG2 and ERG3 gene defects and reduced ergosterol content [59] [Table/Fig-4].
Showing mechanism of antifungal drug resistance to polyenes.
Resistance to flucytosine (5-flurocytosine): Resistance to 5-flurocytosine has been reported in around 10% of C.albicans cases. The drug flurocytosine acts by inhibition of fungal protein synthesis and nucleic acid synthesis. Fungal cytosine permease takes the drug inside the cell. Flurocytosine is first deaminated to 5 fluro uracil and then split into 5 flurodeoxyuradine monophosphate and 5 flurouridylic acid and then phosphorylates into 5 flurouracil triphosphate, which is catalysed by uracil phosphoribosyltransferase. DNA synthesis is inhibited by 5 flurodeoxyuridine monophosphate by inhibiting thymidine synthetase. Protein synthesis is inhibited by 5 flurouracil triphosphate by incorporating into RNA [59].
The loss of the enzyme uridine monophosphate pyrophosphorylase is found to be one of the important causes of resistance to 5 Fluorouracil [63]. It has also been suggested that defect in cytosine deaminase activity may lead to primary resistance and decrease in uracil phosphoribosyltransferase activity which may lead to secondary resistance [64]. In addition, any loss of permease activity may also cause resistance to 5 Fluorouracil [59] [Table/Fig-5].
Mechanism of drug resistance to flucytosine.
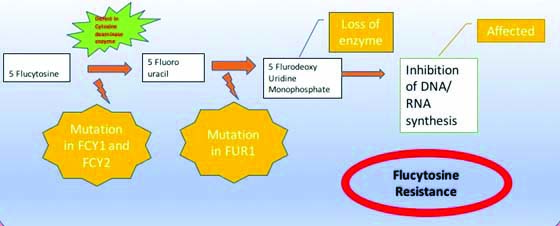
Monotherapy may lead to development of resistance to 5 Fluorouracil. Interestingly, it has been found that when 5 Fluorouracil is given in combination with amphotericin B, the occurrence of drug resistance in C.albicans strains has been reduced. Acquired resistance may be due to failure in conversion of 5-fluorocytosine into 5-fluorouracil triphosphate and 5-flurodeoxyuridinemonophosphate or due to loss of feedback control of pyrimidine biosynthesis. Intrinsic resistance to 5 Fluorouracil may be due to deficiency of enzymes involved in the metabolism of the 5 Fluorouracil pathway.
An overview of various antifungal drugs along with their mechanism of action and drug resistance mechanisms are shown in [Table/Fig-6].
Mechanisms of antifungal drug resistance.
| Antifungal class of drugs | Examples | Mode of action | Mechanism of drug resistance | Molecular basis of Resistance | Final change that leads to resistance | References |
|---|
| Azoles | MiconazoleClotrimazoleKetoconazoleFluconazoleItraconazoleVoriconazole | Inhibitors of lanosterol14-α-deme-thylase | Decreased binding affinity by the drug to lanosterol 14-α-demethylaseIncrease in the concentration levels of lanosterol 14-α-demethylase,Upregulation of drug transporters and alterations in ergosterol biosynthetic pathway | 1.Modifications in the ERG11 gene by: a) Point mutationsb) Up regulation | 1.a) Reduced drug affinity for the target enzyme b) Overexpression Increased ergosterol synthesis | [35][32,34] |
| 2. Alterations in ergosterol biosynthetic pathway | 2. Production of various sterols supporting growth; cross-resistance to other azoles and AmB | [32,33] |
| 3. verexpresssion of CDRs and MDR genes encoding efflux pumps | 3. Reduced drug accumulation in the cell | [30,39,41] |
| Echino-candins | CaspofunginMicafunginAnidulafungin | Inhibitors of(1,3)-α-D-glucan synthase | Decrease in the glycan synthesis processing | 1. Point mutation in FKS subunits of enzyme β(1,3) D-glucan synthase | 1. Decrease in susceptibility to echinocandins | [42] |
| 2. Mutation in hotspot region | 2. Increase in Minimum inhibitory concentration (MIC), reduced β(1,3)D-glucan synthase sensitivity, and cross-resistance among the echinocandins. | [43-45] |
| Polyenes | NystatinAmphotericin B | Bind to ergosterol | Decrease in the ergosterol content in the cell | 1.inhibition ofergosterol synthesis | 1. Decrease in ergosterol levels, replacement of ergosterols withother biosynthetic precursors | [59,60] |
| 2. Masking of the ergosterol in the cell membrane | 2. Nonavailability for binding with the polyenes | [61] |
| 3.Point mutations in ERG3 and ERG6 | 3. Defect in enzymes involved in the synthesis of ergosterol | [59] |
| Nucleoside analogues | Flucytosine | Inhibitor of DNA/RNA synthesis | loss of the enzyme uridine monophosphate pyropho--sphorylase,defect in cytosine deaminase activity,decrease in uracil phosphoribo-syltran-sferase activity,loss of permease activity | 1.Point mutations in FCY1, FCY2, FUR1 | 1. Deficiency in the enzymes necessary for cellular transport and uptake of 5-FC or for its metabolism | [72] |
| 2. Increased synthesis of pyrimidines | 2. It competes with the fluorinated antimetabolites of 5-FC and thus diminish its antimycotic activity. | [72] |
Antifungal resistance and its clinical impact: The resistance exhibited by the Candida species is found to be associated with rise in MICs leading to poor prognosis of the patients, high management costs, etc. It has been reported that the clinical outcome of patients infected with Candida species is significantly poorer in cases of resistant MICs for fluconazole and voriconazole when compared with the susceptible MICs [31]. Few case reports also describe the resistance to echinocandins is associated with high MICs and poor clinical outcomes [65-67]. Various studies have reported breakthrough infections in bone marrow and solid organ transplant patients on fluconazole prophylaxis and epifunginprophylaxis [68,69]. Invasive Candidiasis can also lead to prolonged hospital stay and high management costs due to difficulty in diagnosis and identification of resistant strains [70,71]. However, sparse data is available on the financial impact of resistant Candidal infections.
Conclusion
Infections caused by Candida species is one of the major clinical threats in immunocompromised patients. Although, many antifungal drugs are currently in clinical practice, further novel antifungal drugs and new drug targets are in need due to the fear of antifungal drug resistance and re-emergence of infections. It has become imperative to understand the molecular mechanisms of drug resistance to combat the multidrug resistant fungal infections. Recently, many strategies are being implemented for the prevention of emergence of drug resistance in Candidal infections. Aggressive surveillance of resistance and development of appropriate protocol and guidelines for antifungal drug therapy is the major need today. Antifungal susceptibility testing in all cases is recommended for effective management of Candidal infections. Furthermore molecular, genetic and biological research is necessary to understand the underlying molecular drug resistance mechanisms in antifungal therapy and to discover newer antifungal drugs with high efficacy. Also, there is a need for integration of different disciplines in establishing the management protocol for Candidal infections. Furthermore, exploiting the diagnostic methods including the molecular techniques facilitates effective clinical management to reduce the morbidity and mortality due to candidal infections.
[1]. Ferreira C, Silva S, Faria-Oliveira F, Pinho E, Henriques M, Lucas C, Candida albicans virulence and drug-resistance requires the O-acyltransferase Gup1pBMC Microbiology 2010 10:23810.1186/1471-2180-10-23820843317 [Google Scholar] [CrossRef] [PubMed]
[2]. Spampinato C, Leonardi D, Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agentsBio Med Research International 2013 2013:20423710.1155/2013/20423723878798 [Google Scholar] [CrossRef] [PubMed]
[3]. Arendrup MC, Epidemiology of invasive candidiasisCurr Opin Crit Care 2010 16:445-52.10.1097/MCC.0b013e32833e84d220711075 [Google Scholar] [CrossRef] [PubMed]
[4]. Chow JK, Golan Y, Ruthazer R, Karchmer AW, Carmeli Y, Lichtenberg D, Factors associated with candidemia caused by non-albicans Candida species versus Candida albicans in the intensive care unitClin Infect Dis 2008 46:1206-13.10.1086/52943518444857 [Google Scholar] [CrossRef] [PubMed]
[5]. Samonis G, Kofteridis DP, Saloustros E, Giannopoulou KP, Ntziora F, Christidou A, Candida albicans versus non-albicans bloodstream infection in patients in a tertiary hospital: an analysis of microbiological dataScand J Infect Dis 2008 40:414-19.10.1080/0036554070176565718418802 [Google Scholar] [CrossRef] [PubMed]
[6]. Perfect JR, Schell WA, The new fungal opportunists are comingClin Infect Dis 1996 22(suppl 2):S112-18.10.1093/clinids/22.Supplement_2.S1128722837 [Google Scholar] [CrossRef] [PubMed]
[7]. Pfaller MA, Nosocomial candidiasis. Emerging species, reservoirs and modes of transmissionClin Infect Diseases 1996 22(Suppl 2):S89-94.10.1093/clinids/22.Supplement_2.S898722834 [Google Scholar] [CrossRef] [PubMed]
[8]. Cannon RD, Lamping E, Holmes AR, Niimi K, Tanabe K, Niimi M, Candida albicans drug resistance-another way to cope with stressMicrobiology 2007 153:3211-17.10.1099/mic.0.2007/010405-017906120 [Google Scholar] [CrossRef] [PubMed]
[9]. Cowen LE, Anderson JB, Kohn LM, Evolution of drug resistance in Candida albicansAnnu Rev Microbiol 2002 56:139-65.10.1146/annurev.micro.56.012302.16090712142485 [Google Scholar] [CrossRef] [PubMed]
[10]. Yapar N, Epidemiology and risk factors for invasive candidiasisTher Clin Risk Manag 2014 10:95-105.10.2147/TCRM.S4016024611015 [Google Scholar] [CrossRef] [PubMed]
[11]. Pfaller MA, Antifungal drug resistance: mechanisms, epidemiology and consequences for treatmentAm J Med 2012 125(1 suppl):S3-13.10.1016/j.amjmed.2011.11.00122196207 [Google Scholar] [CrossRef] [PubMed]
[12]. Kaur R, Goyal R, Dhakad MS, Bhalla P, Kumar R, Epidemiology and virulence determinants including biofilm profile of candida infections in an ICU in a tertiary hospital in IndiaJournal of Mycology 2014 2014:30349110.1155/2014/303491 [Google Scholar] [CrossRef]
[13]. Loeffler J, Stevens DA, Antifungal drug resistanceClinical Infectious Diseases 2003 36(Suppl 1):S31-41.10.1086/34465812516028 [Google Scholar] [CrossRef] [PubMed]
[14]. Bongomin F, Gago S, Oladele RO, Denning DW, Global and multi-national prevalence of fungal diseases-estimate precisionJournal of Fungi 2017 3(4):5710.3390/jof304005729371573 [Google Scholar] [CrossRef] [PubMed]
[15]. Giri S, Kindo AJ, A review of candida species causing blood stream infectionIndian J Med Microbiol 2012 30(3):270-78.10.4103/0255-0857.9948422885191 [Google Scholar] [CrossRef] [PubMed]
[16]. Kumar CP, Sundararajan T, Menon T, Venkatadesikalu M, Candidiosis in children with onco-hematological studies in Chennai, South IndiaJpn J Infect Dis 2005 58:218-21. [Google Scholar]
[17]. Sahni V, Agarwal SK, Singh NP, Anuradha S, Sikdar S, Wadhwa A, Candidemia-An Under-recognized nosocomial infection in Indian HospitalsJ Assoc Physicians India 2005 53:607-11. [Google Scholar]
[18]. Kothari A, Sagar V, Epidemiology of candida bloodstream infections in a tertiary care institute in IndiaIndian J Med Microbiol 2008 27(2):171-72.10.4103/0255-0857.4944019384050 [Google Scholar] [CrossRef] [PubMed]
[19]. Xess I, Jain N, Hasan F, Mandal P, Banerjee U, Epidemiology of candidemia in a tertiary care centre of North India: 5-Year StudyInfection 2007 35:256-59.10.1007/s15010-007-6144-617646917 [Google Scholar] [CrossRef] [PubMed]
[20]. Goel N, Ranjan PK, Agarwal R, Chaudhary U, Sanjeev N, Emergence of nonalbicans Candida in neonatal septicemia and antifungal susceptibility: Experience from a tertiary care centreJ Lab Physicians 2009 1:53-55.10.4103/0974-2727.5969921938250 [Google Scholar] [CrossRef] [PubMed]
[21]. Tritipwanit K, Chindamporn A, Suankratay C, Epidemiology of Candidemia at King Chulalongkorn Memorial Hospital, ThailandJ Infect Dis Antimicrob Agents 2005 22(2):59-69. [Google Scholar]
[22]. Verma AK, Prasad KN, Singh M, Dixit AK, Ayyagari A, Candidaemia in patients of a tertiary health care hospital from north IndiaIndian J Med Res 2003 117:122-28. [Google Scholar]
[23]. Denning DW, Hope WW, Therapy for fungal diseases: Opportunities and prioritiesTrends Microbiol 2010 18(5):195-204.10.1016/j.tim.2010.02.00420207544 [Google Scholar] [CrossRef] [PubMed]
[24]. Kathiravan MK, Salake AB, Chothe AS, Dudhe PB, Watode RP, Mukta MS, The biology and chemistry of antifungal agents: a reviewBioorganic & Medicinal Chemistry 2012 20:5678-98.10.1016/j.bmc.2012.04.04522902032 [Google Scholar] [CrossRef] [PubMed]
[25]. Hay R, “Antifungal drugs,”in European Handbook of Dermatological Treatments, Katsambas A and Lotti T, Eds. 2003 Berlin, GermanySpringer:700-10.10.1007/978-3-662-07131-1_122 [Google Scholar] [CrossRef]
[26]. Hof H, A new, broad-spectrum azole antifungal: posaconazole-mechanisms of action and resistance, spectrum of activityMycoses 2006 49(1):02-06.10.1111/j.1439-0507.2006.01295.x16961575 [Google Scholar] [CrossRef] [PubMed]
[27]. Mora-Duarte J, Betts R, Rotstein C, Colombo AL, Thompson-Moya L, Smietana J, Comparison of caspofungin and amphotericin B for invasive candidiasisThe New England Journal of Medicine 2002 347(25):2020-29.10.1056/NEJMoa02158512490683 [Google Scholar] [CrossRef] [PubMed]
[28]. de Wet N, Llanos-Cuentas A, Suleiman J, Baraldi E, Krantz EF, Della Negra M, A randomized, double-blind, parallel-group, dose-response study of micafungin compared with fluconazole for the treatment of esophageal candidiasis in HIV-positive patientsClin Infect Dis 2004 39(6):842-49.10.1086/42337715472817 [Google Scholar] [CrossRef] [PubMed]
[29]. Sanglard D, Coste A, Ferrari S, Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulationFEMS Yeast Research 2009 9(7):1029-50.10.1111/j.1567-1364.2009.00578.x19799636 [Google Scholar] [CrossRef] [PubMed]
[30]. Kanafani ZR, Perfect JR, Resistance to antifungal agents: mechanisms and clinical impactClin Infect Dis 2008 46:120-28.10.1086/52407118171227 [Google Scholar] [CrossRef] [PubMed]
[31]. Tew GN, Clements D, Tang H, Arnt L, Scott RW, Antimicrobial activity of an abiotic host defense peptide mimicBiochim Biophys Acta 2006 1758(9):1387-92.10.1016/j.bbamem.2006.03.00116626628 [Google Scholar] [CrossRef] [PubMed]
[32]. Morschhauser J, The genetic basis of fluconazole resistance development in Candida albicansBiochim Biophys Acta 2002 1587(2-3):240-48.10.1016/S0925-4439(02)00087-X [Google Scholar] [CrossRef]
[33]. Mishra NN, Prasad T, Sharma N, Payasi A, Prasad R, Gupta DK, Pathogenecity and drug resistance in Candida albicans and other yeast species A reviewActa Microbial Immunol Hung 2007 54(3):201-35.10.1556/AMicr.54.2007.3.117896473 [Google Scholar] [CrossRef] [PubMed]
[34]. White TC, Holleman S, Dy F, Mirels LF, Stevens DA, Resistance mechanisms in clinical isolates of Candida albicansAntimicrob Agents Chemother 2002 46:1704-13.10.1128/AAC.46.6.1704-1713.200212019079 [Google Scholar] [CrossRef] [PubMed]
[35]. Löffler J, Kelly SL, Hebart H, Schumacher U, Lass-Flörl C, Einsele H, Molecular analysis of cyp51 from fluconazole-resistant Candida albicans strainsFEMS Microbiol Lett 1997 151:263-68.10.1016/S0378-1097(97)00172-9 [Google Scholar] [CrossRef]
[36]. Orozco AS, Higginbotham LM, Hitchcock CA, Parkinson T, Falconer D, Ibrahim AS, Mechanism of fluconazole resistance in Candida kruseiAntimicrob Agents Chemother 1998 42:2645-69.10.1128/AAC.42.10.26459756770 [Google Scholar] [CrossRef] [PubMed]
[37]. White TC, The presence of an R- 467 K amino acid substitution and loss of alleic variation correlate with an azole-resistant lanosterol 14 α demethylase in Candida albicansAntimicrob Agents Chemother 1997 41:1488-94.10.1128/AAC.41.7.14889210671 [Google Scholar] [CrossRef] [PubMed]
[38]. Kelly SL, Lamb DC, Kelly DE, Manning NJ, Loeffler J, Hebart H, Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol delta5,6-desaturationFEBS Lett 1997 400:80-82.10.1016/S0014-5793(96)01360-9 [Google Scholar] [CrossRef]
[39]. Vanden Bossche H, Warnock DW, Dupont B, Kerridge D, Sen Gupta S, Improvisi L, Mechanisms and clinical impact of antifungal drug resistanceJ Med Vet Mycol 1994 32(suppl 1):189-202.10.1080/02681219480000821 [Google Scholar] [CrossRef]
[40]. Maebashi K, Niimi M, Kudoh M, Fischer FJ, Makimura K, Niimi K, Mechanisms of fluconazole resistance in Candida albicans isolates from Japanese AIDS patientsJ antimicrob Chemother 2001 47:527-36.10.1093/jac/47.5.52711328762 [Google Scholar] [CrossRef] [PubMed]
[41]. Vanden Bossche H, Dromer F, Improvisi I, Lozano-Chiu M, Rex JH, Sanglard D, Antifungal drug resistance in pathogenic fungiMed Mycol 1998 36(Suppl 1):119-28. [Google Scholar]
[42]. Perlin DS, Resistance to echinocandin-class antifungal drugsDrug Resist Updat 2007 10:121-30.10.1016/j.drup.2007.04.00217569573 [Google Scholar] [CrossRef] [PubMed]
[43]. Beyda ND, Lewis RE, Garey KW, Echinocandin resistance in Candida species: mechanisms of reduced susceptibility and therapeutic approachesAnn Pharmacother 2012 46:1086-96.10.1345/aph.1R02022811350 [Google Scholar] [CrossRef] [PubMed]
[44]. Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolatesAntimicrob Agents Chemother 2005 49:3264-73.10.1128/AAC.49.8.3264-3273.200516048935 [Google Scholar] [CrossRef] [PubMed]
[45]. Garcia-Effron G, Park S, Perlin DS, Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpointsAntimicrob Agents Chemother 2009 53(1):112-22.10.1128/AAC.01162-0818955538 [Google Scholar] [CrossRef] [PubMed]
[46]. Garcia-Effron G, Kontoyiannis DP, Lewis RE, Perlin DS, Caspofungin- resistant Candida tropicalis strains causing breakthrough fungemia in patients at high risk for hematologic malignanciesAntimicrob Agents Chemother 2008 52:4181-83.10.1128/AAC.00802-0818794386 [Google Scholar] [CrossRef] [PubMed]
[47]. Garcia-Effron G, Lee S, Park S, Cleary JD, Perlin DS, Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: implication for the existing susceptibility breakpointAntimicrob Agents Chemother 2009 53:3690-99.10.1128/AAC.00443-0919546367 [Google Scholar] [CrossRef] [PubMed]
[48]. Katiyar S, Pfaller M, Edlind T, Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibilityAntimicrob Agents Chemother 2006 50(8):2892-94.10.1128/AAC.00349-0616870797 [Google Scholar] [CrossRef] [PubMed]
[49]. Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteriaDrug Resist Updat 2011 14:164-76.10.1016/j.drup.2011.01.00421353623 [Google Scholar] [CrossRef] [PubMed]
[50]. Sanguinetti M, Posteraro B, Lass-Flörl C, Antifungal drug resistance among Candida species: mechanisms and clinical impactMycoses 2015 58(Suppl. 2):02-13.10.1111/myc.1233026033251 [Google Scholar] [CrossRef] [PubMed]
[51]. Munro CA, Selvaggini S, de Bruijn I, Walker L, Lenardon MD, Gerssen B, The PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin synthesis in Candida albicansMol Microbiol 2007 63:1399-413.10.1111/j.1365-2958.2007.05588.x17302816 [Google Scholar] [CrossRef] [PubMed]
[52]. Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, Gow NA, Stimulation of chitin synthesis rescues Candida albicans from echinocandinsPLoS Pathog 2008 4:e100004010.1371/journal.ppat.100004018389063 [Google Scholar] [CrossRef] [PubMed]
[53]. Stevens DA, Espiritu M, Parmar R, Paradoxical effect of caspofungin: reduced activity against Candida albicans at high drug concentrationsAntimicrob Agents Chemother 2004 48:3407-11.10.1128/AAC.48.9.3407-3411.200415328104 [Google Scholar] [CrossRef] [PubMed]
[54]. Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H, Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofunginAntimicrob Agents Chemother 2006 50:3160-61.10.1128/AAC.00563-0616940118 [Google Scholar] [CrossRef] [PubMed]
[55]. Chamilos G, Lewis RE, Albert N, Kontoyiannis DP, Paradoxical effect of echinocandins across Candida species in vitro: evidence for echinocandin-specific and Candida species-related differencesAntimicrob Agents Chemother 2007 51:2257-59.10.1128/AAC.00095-0717438060 [Google Scholar] [CrossRef] [PubMed]
[56]. Shields RK, Nguyen MH, Du C, Press E, Cheng S, Clancy CJ, Paradoxical effect of caspofungin against Candida bloodstream isolates is mediated by multiple pathways but eliminated in human serumAntimicrob Agents Chemother 2011 55:2641-47.10.1128/AAC.00999-1021422223 [Google Scholar] [CrossRef] [PubMed]
[57]. Ellis D, Amphotericin B: spectrum and resistanceJ Antimicrob Chemother 2002 49:07-10.10.1093/jac/49.suppl_1.711801575 [Google Scholar] [CrossRef] [PubMed]
[58]. Laniado-Laboroın R, Cabrales-Vargas MN, Amphotericin B: side effects and toxicityRev lberoam deMicol 2009 26(4):223-27.10.1016/j.riam.2009.06.00319836985 [Google Scholar] [CrossRef] [PubMed]
[59]. Arikan S, Rex JH, “Resistance to antifungal agents,”in Topley and Wilson’s Microbiology and Microbial Infections: Medical Mycology, 10th Edn, edsMerz W.G., Hay R.J., editors 2005 LondonHodder Arnold:168-181. [Google Scholar]
[60]. Peyron F, Favel A, Calaf R, Michel-Nguyen A, Bonaly R, Coulon J, Sterol and Fatty acid composition of Candida lusitaniae clinical isolatesAntimicrob Agents Chemother 2002 46:531-33.10.1128/AAC.46.2.531-533.200211796371 [Google Scholar] [CrossRef] [PubMed]
[61]. Ghannoum MA, Rice LB, Antifungal agents. Mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistanceClin Microbiol Rev 1999 12:501-17.10.1128/CMR.12.4.501 [Google Scholar] [CrossRef]
[62]. Odds FC, Antifungal agents and their use in Candida infectionsIn: Candida and candidosis 1988 2nd edLondonBilliere Tindall:279-313. [Google Scholar]
[63]. Lambert HP, O’Grades FW, Antibiotic and chemotherapy 1992 6th edLondonChurchill- Livingstone:27-37. [Google Scholar]
[64]. Sheikh N, Jahagirdar V, Kothadia S, Nagoba B, Antifungal drug resistance in candida speciesEur J Gen Med 2013 10(4):254-58.10.29333/ejgm/82217 [Google Scholar] [CrossRef]
[65]. Hakki M, Staab JF, Marr KA, Emergence of a Candida krusei isolate with reduced susceptibility to caspofungin during therapyAntimicrob Agents Chemother 2006 50:2522-54.10.1128/AAC.00148-0616801435 [Google Scholar] [CrossRef] [PubMed]
[66]. Miller CD, Lomaestro BW, Park S, Perlin DS, Progressive esophagitis caused by Candida albicans with reduced susceptibility to caspofunginPharmacotherapy 2006 26:877-80.10.1592/phco.26.6.87716716141 [Google Scholar] [CrossRef] [PubMed]
[67]. Baixench MT, Aoun N, Desnos-Ollivier M, Garcia-Hermoso D, Bretagne S, Ramires S, Acquired resistance to echinocandins in Candida albicans: case report and reviewJ Antimicrob Chemother 2007 59:1076-83.10.1093/jac/dkm09517468115 [Google Scholar] [CrossRef] [PubMed]
[68]. Alexander BD, Schell WA, Miller JL, Long GD, Perfect JR, Candida glabratafungemia in transplant patients receiving voriconazole after fluconazoleTransplantation 2005 80:868-71.10.1097/01.tp.0000173771.47698.7b16210978 [Google Scholar] [CrossRef] [PubMed]
[69]. Pfeiffer CD, Garcia-Effron G, Zaas AK, Perfect JR, Perlin DS, Alexander BD, Breakthrough invasive candidiasis in patients on micafunginJ Clin Microbiol 2010 48:2373-80.10.1128/JCM.02390-0920421445 [Google Scholar] [CrossRef] [PubMed]
[70]. Fridkin SK, Candidemia is costly-plain and simpleClin Infect Dis 2005 41:1240-41.10.1086/49693516206096 [Google Scholar] [CrossRef] [PubMed]
[71]. Morgan J, Meltzer MI, Plikaytis BD, Sofair AN, Huie-White S, Wilcox S, Excess mortality, hospital stay, and cost due to candidemia: a case-control study using data from population-based candidemia surveillanceInfect Control Hosp Epidemiol 2005 26:540-47.10.1086/50258116018429 [Google Scholar] [CrossRef] [PubMed]
[72]. Vermes A, Guchelaar HJ, Dankert J, Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactionsJ Antimicrob Chemother 2000 46(2):171-79.10.1093/jac/46.2.17110933638 [Google Scholar] [CrossRef] [PubMed]