Case Report
A five-year-old girl was referred to the endocrinology and metabolism clinic of Imam Hussein Children’s Hospital, the referral hospital for paediatric disease affiliated to Isfahan University of Medical Sciences for evaluation of growth retardation. She was referred because of growth retardation and short stature.
The patient was the second child of consanguineous parents born by elective cesarean section at 38 weeks of gestation in 2009. Prenatal ultrasonography diagnosed intrauterine growth restriction, but no other abnormality or malformation was detected. Birth weight, height and head circumference were 2200 gram (<5th percentile), 46 cm (<5th percentile) and 34 cm (25th percentile), respectively. Neonatal screening tests were normal. The patient had normal development but growth rate was slower than normal. In order to determine development and intelligence of the patient, the "Stanford-Binnet" test was performed by a psychiatrist. The results did not indicate mental and developmental retardation. She had no history of any disease or hospital admissions. She received all compulsory vaccines, according to the schedule of Iran national vaccination program. She had no history of recurrent infection. Both parents were healthy with no history of any certain disease. Elder sister of the patient died at two years due to nephrotic syndrome.
The patient was clinically examined by a paediatric endocrinologist. According to the age of the patient, her development was normal. Weight and height of the patient were 11.3 kg (below 5% percentile) and 86 cm (below 5% percentile, height SDS -5.7), respectively. Other anthropometrics findings including sitting height/leg length and upper segment/lower segment ratio were 0.8 and 0.83 respectively. She had short neck, short trunk, triangular face and a broad nasal bridge [Table/Fig-1a].
The physical findings of the patient.
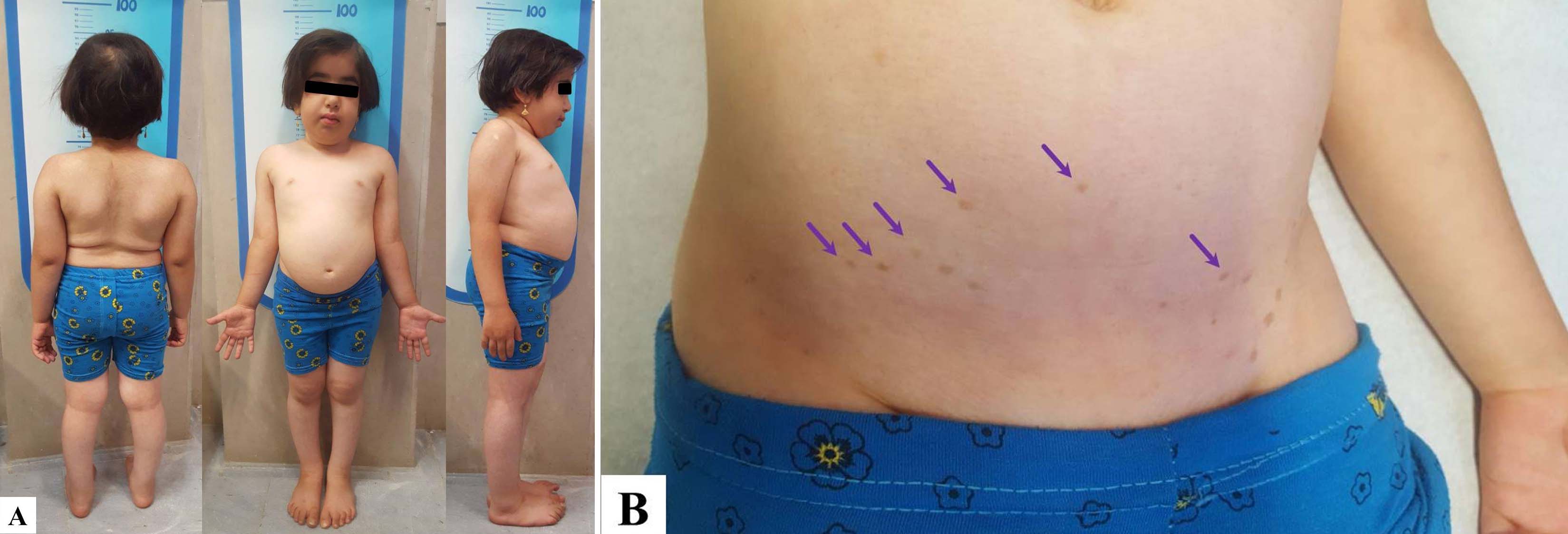
Systolic and diastolic blood pressures of the patient were 105 and 65 mmHg, respectively. Puberty stage was Tanner stage 1. There were hyperpigmented macules in the lower abdominal region [Table/Fig-1b]. Physical examination of thyroid was normal, and there was no abnormality on the chest.
Levels of serum glucose, electrolytes, and thyroid function were normal. Renal function test and complete blood count were normal. A 24 hours urine analysis and stool exam was negative. Serum immunoglobulin levels (i.e., IgA, IgM and IgG) were normal. Considering the patient’s disproportionate growth and dermatologic findings, genetic study was performed for the patient and her parents. Molecular genetic testing indicated biallelic pathogenic variants in SWI/SNF2-related, Matrix-associated, Actin-dependent Regulator of Chromatin, subfamily A-Like 1 (SMARCAL1). Both parents were heterogeneous for the SMARCAL1 variants.
Considering the result of genetic testing, the diagnosis of SIOD was confirmed. Following diagnosis of SIOD, the patient was referred to a paediatric nephrologist for evaluation. Serum creatinine and urea level were normal. Creatinine clearance was normal and she had no proteinuria. She had no anaemia, neutropenia, lymphopenia and thrombocytopenia. Immunologic tests were normal. The patient was referred to dentist, ophthalmologist, paediatric neurologist and orthopedist for further evaluation. There were no findings during their evaluations. The patient was recommended to refer to the paediatric endocrinology clinic for periodic follow up.
During the periodic follow up, at age of 6.5 years, results of renal function test indicated proteinuria. The results of her biochemical tests showed in [Table/Fig-2]. The patient was diagnosed with nephrotic syndrome in December 2016 and underwent treatment with prednisolone10 mg daily. Renal creatinine clearance was 75 mL/min /1.73 m2.
The Results of biochemical tests during the periodic follow up at the age of 6.5 years.
| Test | Value | Reference | Units |
|---|
| Serum albumin | 2.4 | 3.7 - 5.5 | gm/dL |
| Serum creatinine | 0.7 | 0.12 - 1.06 | mg/dL |
| Haemoglobin | 11.2 | 11.5 - 15.5 | gm/dL |
| White blood cells | 6.4 | 5.0 - 14.5 | ×103/μL |
| Platelets | 164 | 150 - 450 | ×103/μL |
| Calcium | 8.3 | 8.8 - 10.1 | mg/dL |
| Phosphorus | 4.9 | 3.1 - 6.3 | mg/dL |
| TSH | 4.2 | 0.5 - 4.5 | mIU/L |
| T4 | 8.6 | 6.4 - 13.3 | μg/dL |
At age of 7.5 years, the patient was referred with severe proteinuria and oedema. Then she was admitted to the hospital for treatment. During the patient’s stay at hospital, hypertension (180/110 mmHg) and seizure developed. Serum albumin and haemoglobin were 2.6 mg/dL and 11 gm/dL, respectively. White blood cells were normal. Prednisolone, albumin and labetalol were administrated. Renal biopsy was performed and the result was Focal and Segmental Glomerulosclerosis (FSGS). Magnetic Resonance Imaging (MRI) of the brain showed hydrocephalus due to sylvius aqueduct stenosis. There was no abnormality in the MRI. In order to control the blood pressure, Spironolactone (12.5 mg/day) was prescribed at the time of the discharge. Considering progressive proteinuria of the patient, cyclosporine was also administrated. During the follow up period (last follow up was in January 2018), the patient had no sign and laboratory finding related to her immune deficiency.
Discussion
Schimke-immuno-osseous dysplasia (SIOD) is a rare multisystem progressive disorder which was first described by Schimke RN et al., in 1971 [1]. SIOD is known as a very rare disorder. Prevalence of this disease have been estimated as less than one per million [2]. It is a pan-ethnic disorder and is not associated with geographic region, ethnicity and gender [3]. The disorder has an autosomal recessive inheritance pattern due to biallelic mutations in SMARCAL1 gene [4]. The protein encoded by this gene is a member of the SWI/SNF family of proteins; which are thought to regulate transcription of certain genes by altering the chromatin structure around those genes.
The main clinical manifestations of SIOD are disproportionate short stature, spondyloepiphyseal dysplasia, dysmorphism, hyperpigmented macules, and progressive renal disease commonly presented by FSGS and signs and symptoms of impaired cellular immunity [5]. SIOD could also manifest by autoimmune diseases, neurological complications such as transient cerebral ischaemia, chronic headache, cancer such as non-Hodgkin’s lymphoma or osteosarcoma, dental malformations, bone marrow aplasia and hypothyroidism [6-8]. It usually present with growth failure and other mentioned disease diagnosed during its follow-up and management period [9].
Base on clinical manifestations and age of disease onset, it is classified in to severe early-onset/infantile and milder late-onset/ juvenile forms. The outcomes of the two forms are early death and survival into adulthood for the mild and severe forms, respectively [9]. In this report we present a case of SIOD from a consanguineous Iranian family. Informed consent was received from the family.
The reported case could be categorized as a mild case of SIOD probably with better survival because she had no thyroid dysfunction, immunodeficiency, recurrent infection or bone marrow failure during follow-up period [10].
Boerkoel CF et al., reported two cases of SIOD (sisters) with consanguineous parents [11]. SIOD in one of them manifested with recurrent herpes labialis and steroid-resistant nephrotic syndrome with hypertension. The second case was presented with hypertension and nephrotic syndrome which progressed to end-stage renal failure. Taha D et al., have reported a case of SIOD from Saudi Arabia whose parents were first cousins. SIOD in this patient represented with fever of unknown origin as a result of B-cell lymphoma. In family history of the patients, he had a brother who died at three years of age and had poor growth rate and features like this one [12]. Recently, Bakr A et al., reported a case on late onset SIOD which manifested with short stature, spondyloepiphyseal dysplasia, FSGS and cellular immune deficiency with parental consanguinity [13]. In the present case, the patient had a sister who died at 2 years of age due to nephrotic syndrome. It could be suggested that the sister had early onset and severe form of SIOD.
A recent study demonstrated that the chance of being affected, asymptomatic carrier and unaffected in siblings of an affected patient is 25%, 50% and 25%, respectively [5]. In addition, some studies indicated that genotype of patients could not predict the severity and outcome of the disease even among families. It is suggested that factors such as oligogenic inheritance, environment and epigenetics could modify the manifestations and outcome of SIOD [14,15].
In the present case, the patient had normal development. Previous studies also indicated that almost all of the SIOD cases had normal development. Reported developmental delays mainly occur in SIOD cases with early recurrent cerebral ischemic events or as a result of chronic illness. The first manifestation of currently reported case of SIOD likewise most of the previously reported cases has been poor growth [9].
Reported mean age of growth failure in SIOD patients is 2 years ranging from 0-13 years [14]. Evidences indicated that the poor growth is not the consequence of renal dysfunction in SIOD patients. The difference between patients with SIOD from those with non-SIOD Chronic Kidney Disease (CKD) is that the leg length reduction is more significant than trunk length reduction. In SIOD patients with CKD, the sitting height/leg-length ratio is less than 0.83 [13,16]. In our reported case the ratio was 0.8.
In the present case, nephrotic syndrome developed 1.5 years after SIOD diagnosis with the FSGS which considered the most common renal pathology in SIOD. Evidences indicated that SIOD related nephropathy mainly develops within five years after its diagnosis and before 12 years of age.
The index patient had hyperpigmented macules on lower abdominal region. Accordingly, most of the patients with SIOD have hyperpigmented macules more commonly on the trunk and less frequently on the face, neck and extremities [9]. Although most of the early onset and severe form of the SIOD usually die at a younger age and those with milder form survive to adulthood, there are also documents which show severity and age of onset could not definitely predict the survival of SIOD [10,17].
The most common causes of death in SIOD in order of frequency are infection, cerebrovascular events, congestive heart failure, pulmonary hypertension and renal failure. There are evidences which demonstrate that the time of renal failure development could predict the disease prognosis as well [5,18]. Other causes of death are organ transplant complication, complications of lymphoproliferative disease, gastrointestinal haemorrhage, bone marrow aplasia and acute restrictive lung disease.
SMARCAL1 deficiency is the cause of SIOD which is characterized by T cell immunodeficiency. Lev A et al., reported that in SIOD patients the thymus function were reduced remarkably and that was correlated with clinical phenotype of these SIOD patients. Thymus dysfunction in a SIOD patient with no clinical signs of immunodeficiency can be considered as a predictor for future immune system dysfunction. Therefore, this can predispose the patient to various infections [19].
Conclusion
The patient had the typical manifestation of SIOD which presented first by growth failure and the renal dysfunction. The most important thing about the case were her parental consanguinity and having a sister with nephrotic syndrome who died early in life. It is recommended that in such cases with poor growth and a suspected family history, one should consider the evaluations regarding SIOD. In addition proper short-interval follow up is recommended for cases with diagnosis of SIOD in order to better evaluation and management of the disease related complication.
[1]. Schimke RN, Horton WA, King CR, Chondroitin-6-sulphaturia, defective cellular immunity, and nephrotic syndromeLancet 1971 2(7733):1088-89.10.1016/S0140-6736(71)90400-428796785 [Google Scholar] [CrossRef] [PubMed]
[2]. Lipska-Zietkiewicz BS, Gellermann J, Boyer O, Gribouval O, Zietkiewicz S, Kari JA, Low renal but high extrarenal phenotype variability in Schimke immuno-osseous dysplasiaPLoS One 2017 12(8):e018092610.1136/jmg.2006.04431316840568 [Google Scholar] [CrossRef] [PubMed]
[3]. Clewing JM, Antalfy BC, Lücke T, Najafian B, Marwedel KM, Hori A, Schimkeimmuno-osseous dysplasia: a clinicopathological correlationJ Med Genet 2007 44(2):122-30.10.1002/pbc.24542 [Google Scholar] [CrossRef]
[4]. Carroll C, Badu-Nkansah A, Hunley T, Baradaran-Heravi A, Cortez D, Frangoul H, SchimkeImmunoosseous Dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 mutation in a childPediatr Blood Cancer 2013 60(9):E88-90.10.1002/pbc.2454223630135 [Google Scholar] [CrossRef] [PubMed]
[5]. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Schimke Immunoosseous Dysplasia 1993-2017 Seattle (WA)University of Washington [Google Scholar]
[6]. Morimoto M, Kérourédan O, Gendronneau M, Shuen C, Baradaran-Heravi A, Asakura Y, Dental abnormalities in Schimkeimmuno-osseous dysplasiaJ Dent Res 2012 91(7 Suppl):29S-37S.10.1177/002203451245029922699664 [Google Scholar] [CrossRef] [PubMed]
[7]. Ludman MD, Cole DE, Crocker JF, Cohen MM Jr, Schimkeimmuno-osseous dysplasia: case report and reviewAm J Med Genet 1991 47(5):793-96.10.1002/ajmg.13204705388267014 [Google Scholar] [CrossRef] [PubMed]
[8]. Saraiva JM, Dinis A, Resende C, Faria E, Gomes C, Correia AJ, Schimkeimmuno-osseous dysplasia: case report and review of 25 patientsJ Med Genet 1999 36(10):786-89.10.1136/jmg.36.10.78610528861 [Google Scholar] [CrossRef] [PubMed]
[9]. Boerkoel CF, O’Neill S, André JL, Benke PJ, Bogdanovíc R, Bulla M, Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literatureEur J Pediatr 2000 159(1-2):01-07.10.1007/s00431005000110653321 [Google Scholar] [CrossRef] [PubMed]
[10]. Lou S, Lamfers P, McGuire N, Boerkoel CF, Longevity in Schimkeimmuno-osseous dysplasiaJ Med Genet 2002 39(12):922-25.10.1136/jmg.39.12.92212471207 [Google Scholar] [CrossRef] [PubMed]
[11]. Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, Mutant chromatin remodeling protein SMARCAL1 causes Schimkeimmuno-osseous dysplasiaNat Genet 2002 30(2):215-20.10.1038/ng82111799392 [Google Scholar] [CrossRef] [PubMed]
[12]. Taha D, Boerkoel CF, Balfe JW, Khalifah M, Sloan EA, Barbar M, Fatal lymphoproliferative disorder in a child with Schimkeimmuno-osseous dysplasiaAm J Med Genet A 2004 131(2):194-99.10.1002/ajmg.a.3035615523612 [Google Scholar] [CrossRef] [PubMed]
[13]. Bakr A, Eid R, Sarhan A, Hammad A, El-Refaey AM, El-Mougy A, Schimke immune-osseous dysplasia: A case reportSaudi J Kidney Dis Transpl 2015 26(5):987-91.10.4103/1319-2442.16458526354575 [Google Scholar] [CrossRef] [PubMed]
[14]. Clewing JM, Fryssira H, Goodman D, Smithson SF, Sloan EA, Lou S, Schimke-immunoosseous dysplasia: suggestions of genetic diversityHum Mutat 2007 28(3):273-83.10.1002/humu.2043217089404 [Google Scholar] [CrossRef] [PubMed]
[15]. Baradaran-Heravi A, Cho KS, Tolhuis B, Sanyal M, Morozova O, Morimoto M, Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expressionHum Mol Genet 2012 21(11):2572-87.10.1093/hmg/dds08322378147 [Google Scholar] [CrossRef] [PubMed]
[16]. Lücke T, Franke D, Clewing JM, Boerkoel CF, Ehrich JH, Das AM, Schimke versus non-Schimke chronic kidney disease: an anthropometric approachPediatrics 2006 118(2):e400-07.10.1542/peds.2005-261416816006 [Google Scholar] [CrossRef] [PubMed]
[17]. Lücke T, Marwedel KM, Kanzelmeyer NK, Hori A, Offner G, Kreipe HH, Generalized atherosclerosis sparing the transplanted kidney in Schimke diseasePediatr Nephrol 2004 19(6):672-75.10.1007/s00467-004-1426-z15054643 [Google Scholar] [CrossRef] [PubMed]
[18]. Baradaran-Heravi A, Lange J, Asakura Y, Cochat P, Massella L, Boerkoel CF, Bone marrow transplantation in Schimkeimmuno-osseous dysplasiaAm J Med Genet A 2013 161A(10):2609-13.10.1002/ajmg.a.3611123950031 [Google Scholar] [CrossRef] [PubMed]
[19]. Lev A, Amariglio N, Levy Y, Spirer Z, Anikster Y, Rechavi G, Molecular assessment of thymic capacities in patients with Schimke immuno-osseous dysplasiaClin Immunol 2009 133(3):375-81.10.1016/j.clim.2009.08.01719796992 [Google Scholar] [CrossRef] [PubMed]