Malignant Peripheral Nerve Sheath Tumour of the Ovary
Dileep Damodaran1, Mayank Gupta2, Nobin Babu Kalappurayil3, KP Kavitha4, Prashanth Parameswaran5
1 Senior Consultant and Head, Department of Surgical Oncology, MVRCCRI, Kozhikode, Kerala, India.
2 Associate Professor, Department of General Pathology, CMC, Vellore, Tamilnadu, India.
3 Consultant, Department of Pathology, MVRCCRI, Kozhikode, Kerala, India.
4 Senior Consultant, Department of Pathology, ASTER MIMS, Kozhikode, Kerala, India.
5 Consultant, Department of Medical Oncology, MVRCCRI, Kozhikode, Kerala, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Nobin Babu Kalappurayil, Kalappurayil, Florican Road, Karaparamba PO, Kozhikode, Kerala-673010, India.
E-mail: nbk251@gmail.com
Malignant Peripheral Nerve Sheath Tumour (MPNST) of the ovary is a rare disease, and we hereby describe this entity in a 51-year-old lady who presented with an ovarian mass and disseminated peritoneal disease. She was treated initially with Neo Adjuvant Chemotherapy (NACT) thinking it to be a surface epithelial ovarian tumour and subsequently underwent interval cyto reduction. The nerve sheath origin was confirmed by Immunohistochemistry (IHC) studies. There are no standard treatment guidelines for this rare tumour of the ovary.
Neo adjuvant chemotherapy, Nestin, Undifferentiated malignancy
Case Report
A 51-year-old post-menopausal lady presented with heaviness and diffuse pain in the abdomen for two months duration. She has two children and her last childbirth was 23 years ago. Her sister had breast carcinoma which was diagnosed at the age of 47 years. There was no personal and family history of neurofibromatosis.
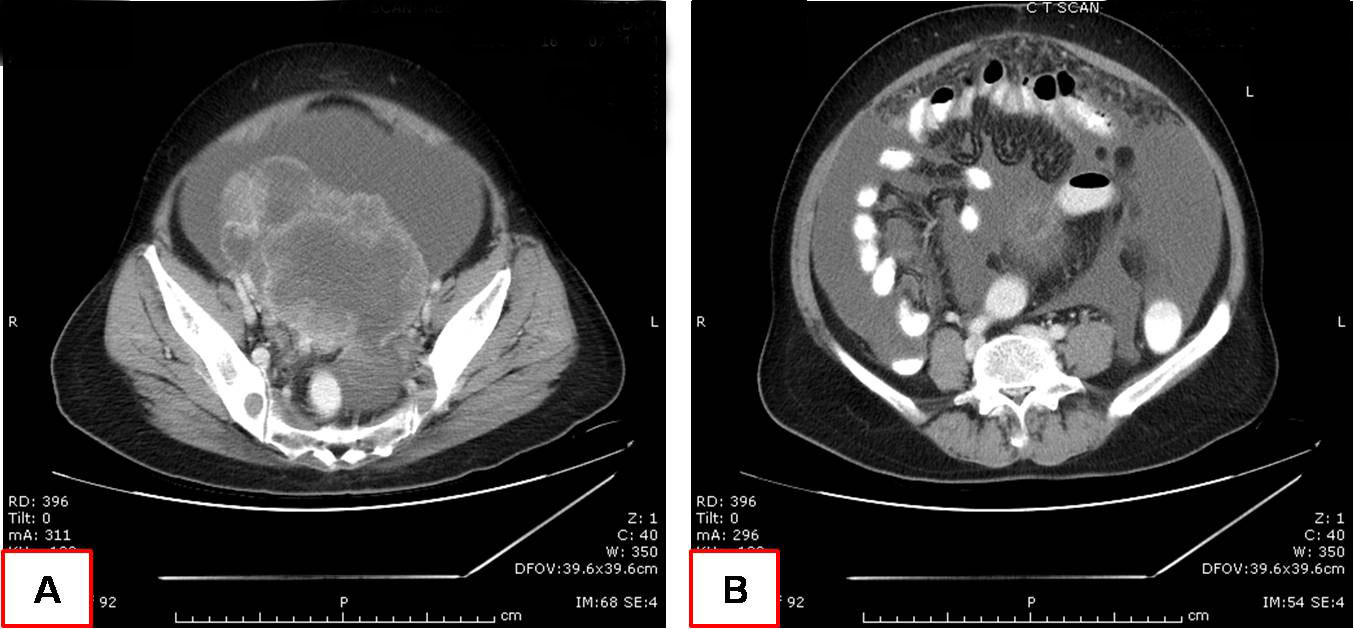
On examination she had distended abdomen, with a 16 week size pelvic mass and ascites. Per vaginal and per speculum examination were unremarkable except for an adnexal mass which was not freely mobile. Digital rectal examination was normal. CT scan of the abdomen and pelvis showed a large adnexal mass of 12.8 X 10.3 X 10.5 cm size with enhancing solid components and septae. Ascites with nodular omental thickening and an aortocaval node of 12mm was also noted [Table/Fig-1]. USG guided fluid cytology and core needle biopsy from the adnexal mass were done. Cytology showed atypical cells suspicious of malignancy and core needle biopsy was reported as poorly differentiated carcinoma. Since the tumour was scanty, IHC markers could not be performed. Serum CA 125 level was 769 Units per ml. As the clinical, radiological and biochemical findings pointed towards a stage III C epithelial ovarian cancer, she was planned for NACT. She underwent three cycles of NACT with paclitaxel and carboplatin.
CT scan of the abdomen and pelvis; A) Large adnexal mass with enhancing solid components and septae; B) Ascites with nodular omental thickening and an aortocaval node.
Subsequent evaluation showed good clinical, radiological and biochemical response. Ascites and nodular omental thickening showed complete resolution in ultrasonography, CA 125 level reached 32.61 units per ml. The size of the adnexal mass remained almost the same as prechemotherapy. She underwent interval cytoreductive procedure wherein an optimal cytoreduction encompassing total abdominal hysterectomy and bilateral salpingo-oophorectomy, bilateral pelvic node dissection and omentectomy were achieved with no evidence of any gross residual disease postoperatively. As per the institutional protocol, an intraperitoneal chemotherapy with cis-diamminedichloro platinum II was given on next postoperative day through an infant feeding tube catheter placed intraoperatively. After the preliminary histopathological examination it was reported as undifferentiated malignancy of ovarian origin. Omentectomy, bilateral pelvic nodes, para aortic node, peritoneal sampling and uterus were reported as free of tumour. Since the pathological findings were not fitting into classical epithelial ovarian carcinoma, the samples were subjected to an in-depth re-evaluation with extended panels of IHC markers.
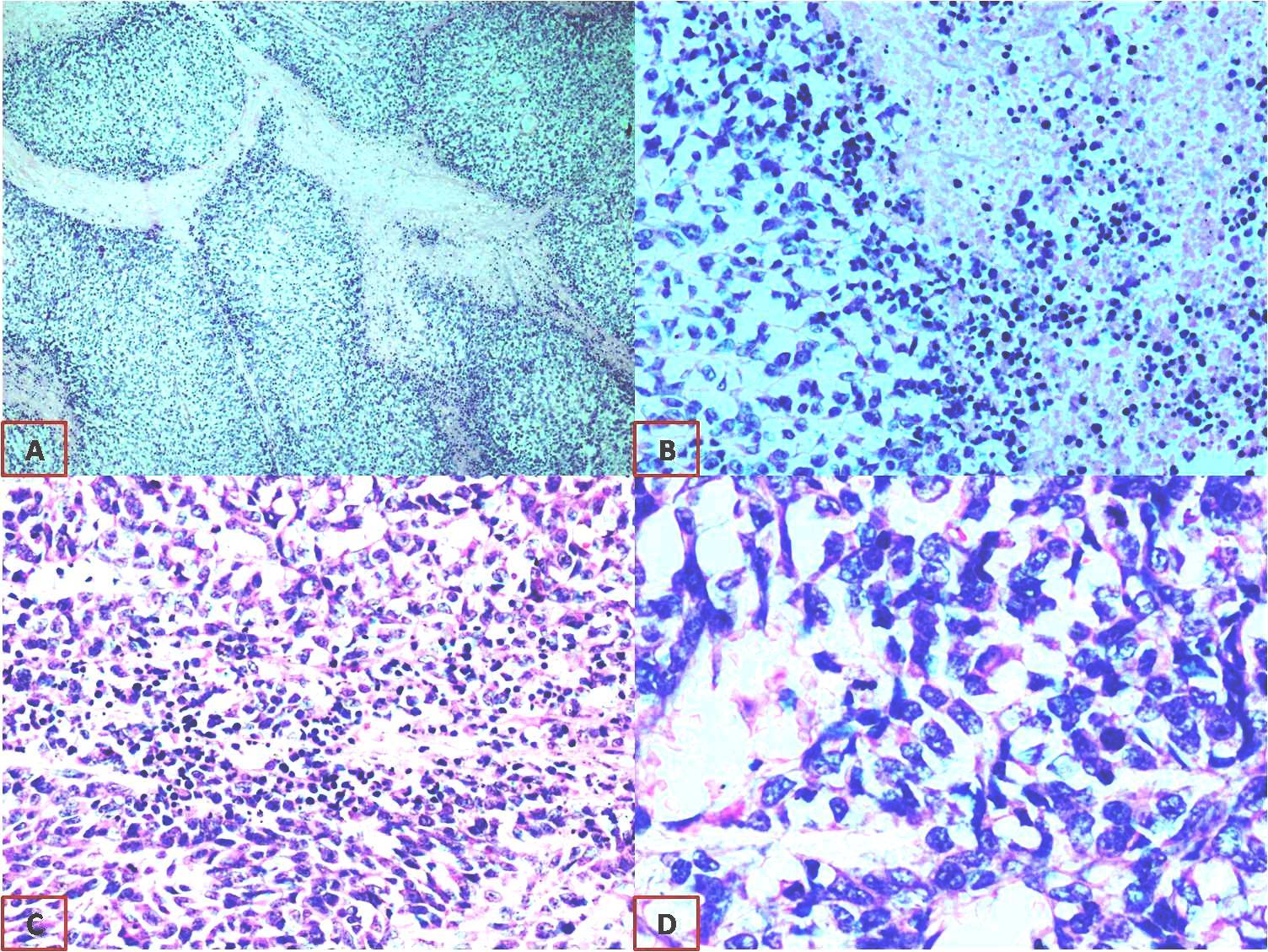
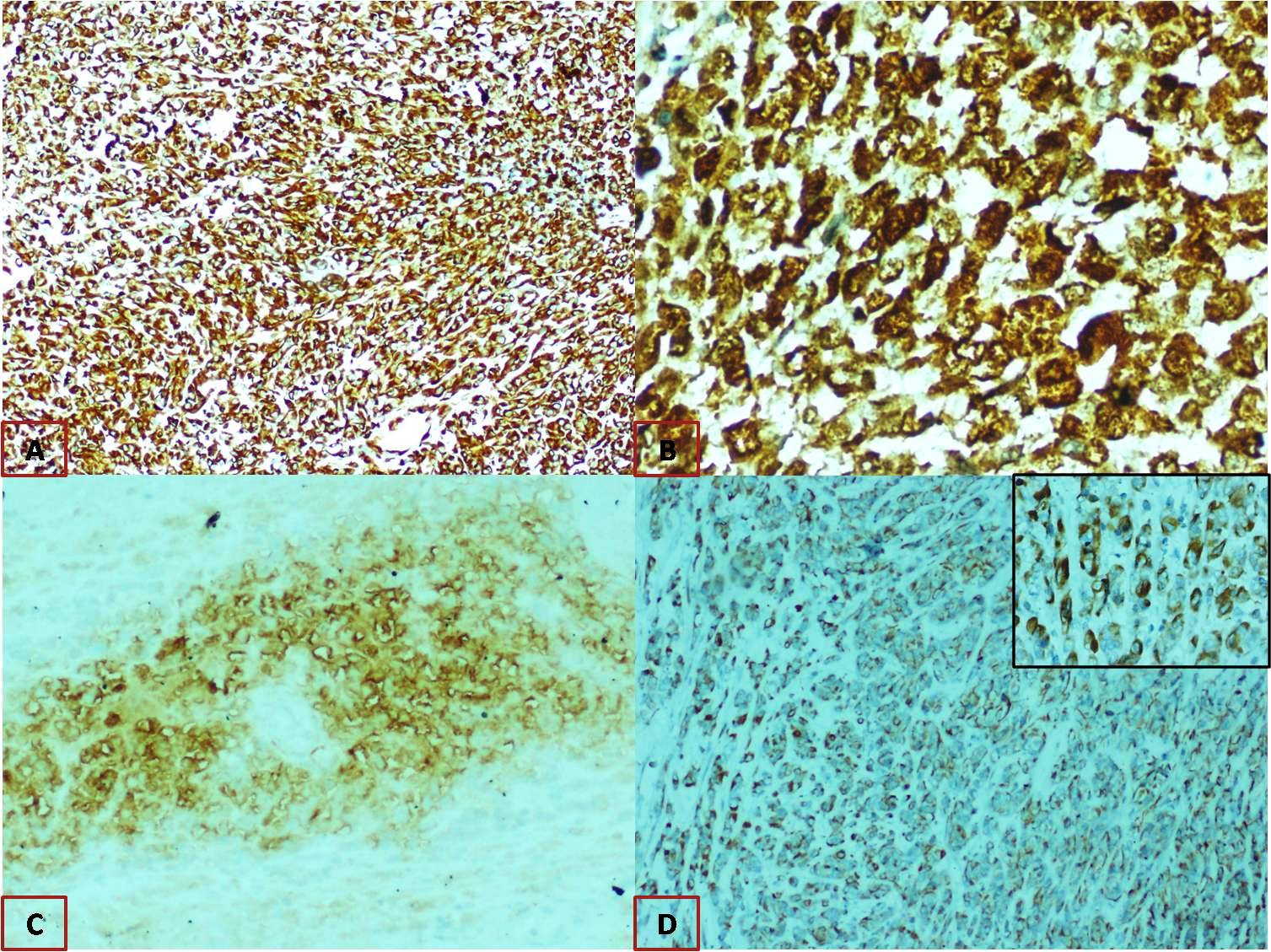
Microscopically, it appeared as solid cellular tumour with plump oval to spindle tumour cells with focal areas of necrosis [Table/Fig-2]. IHC studies revealed diffuse and strong positivity for Vimentin and Nestin, patchy strong positivity for S 100 and focal positivity for CD 57 [Table/Fig-3]. Pancytokeratin and PAX8 were negative, thus epithelial differentiation was ruled out. S100 and CD 57 are neural differentiation markers and nestin being a neuroectodermal stem cell marker. These findings were suggestive of MPNST arising from the ovary. Electron microscopy was done for confirmation, which showed no definite ultra structural features of epithelial differentiation. Few cells showed features suggestive of malignant neural elements but unequivocal evidence of neural or vascular differentiation was not documented. This inconclusive finding for neural differentiation may not be representative of whole tumour since only minimal tissue can be examined by electron microscopy.
A) Tumour arranged as lobules {H&E stain (40X)}; B) Focus displaying tumour with area of necrosis {H&E stain (100X)}; C) Plump oval to spindle tumour cells {H&E stain (100X)}; D) Tumour cells {H&E stains (400X)}.
Immunohistochemistry A) Diffuse strong cytoplasm positivity for vimentin, 40x magnification; B) Patchy strong nuclear and cytoplasmic positivity for S100, 400x. magnification; C) Focal positivity for CD57, 100x magnification; D) Diffuse strong cytoplasmic positivity for nestin, 100x magnification {inset: 400x magnification}.
She completed her scheduled protocol chemotherapy and remains disease free at one and half year follow up. CT scan of abdomen and pelvis showed no evidence of recurrence and CA 125 remains normalized at 6 units per ml. Germline mutational analysis for NF 1/NF 2 was also done, which was negative.
Discussion
The MPNST is a rare soft tissue sarcoma. The incidence among general population is around 0.001% [1]. The most common sites being the extremities, trunk, head and neck region and less commonly it involves the retroperitoneum and intra-abdominal organs [1,2]. In our patient, the diagnosis of MPNST was made post- interval cytoreductive surgery. Diagnosis was suggested based on light microscopy studies and subsequently confirmed by IHC for neural differentiation. Nestin expression has emerged recently as a superior marker for MPNST, was strongly positive in our case [3]. Presence of markers of neural differentiation like S 100 and CD 57 also substantiated the histopathological diagnosis of MPNST [4]. Recently described immunohistochemical marker, H3K27me3 (i.e., trimethylation of histone 3 at lysine 27) can be used to differentiate benign from malignant peripheral nerve sheath tumours. Expression of H3K27me3 is lost in ~30% of low grade, ~60% of intermediate grade, and ~80% of high grade MPNST, whereas retained in benign peripheral nerve sheath tumours [5].
Electron microscopic confirmation of MPNST is difficult but unequivocal findings of neural elements like slender overlapping cytoplasmic processes enveloping other cell bodies, flocculent, granular, material coursing parallel to the plasma membranes and absence of fine intracytoplasmic filaments are confirmatory [6]. An unequivocal evidence of neuronal differentiation could not be obtained in our case. This could possibly be due to chemotherapy changes and non representative sample. There are prior reports which indicate that electron microscopic confirmation of MPNST could be extremely difficult [7].
There are several hypothesis regarding possible origin of a nerve sheath tumour from ovary. Some believe it arises from the small nerves located at the hilum of the ovary which may be nerve plexuses traversing along with the ovarian vessels. The nerves of sympathetic origin may traverse within the ovarian stroma, which might explain the intraparenchymal origin of such tumours [8]. Another less probable explanation is the monodermal differentiation of the germ cell tumour of the ovary or as a predominant element in teratoma [9].
Due to rarity of this tumour and paucity of literature there are no definite treatment guidelines [10-14]. Various chemotherapy regimens and radiotherapy to abdomen has been employed with mixed outcome. We used paclitaxel and carboplatin as neoadjuvant therapy followed by same regimen as adjuvant therapy, since the patient responded well to the agents initially. Radiotherapy to the abdomen, extrapolating the data from soft tissue sarcoma of the extremities was not contemplated in this patient as it carried high morbidity and no data to support its use was evident. Our patient is on close follow up, is doing fine, without any recurrence, one and half years after her surgery.
Conclusion
MPNST of ovary is an extremely rare tumour. As in the literature, worldwide, this is the fourth case being reported and the first one from Asia. Histopathological suspicion and detailed IHC studies will help in diagnosing this rare disease. Electron microscopic features will confirm the neural differentiation, however it may be difficult. There are no standard treatment guidelines for this rare tumour of the ovary.
[1]. Yaga US, Shivakumar R, Kumar MA, Sathyaprakash Malignant peripheral nerve sheath tumour: A rarityIndian J Dent 2015 6(1):53-56.10.4103/0975-962X.15171225767362 [Google Scholar] [CrossRef] [PubMed]
[2]. László A, Ivaskevics K, Sápi Z, Malignant epithelioid ovarian schwannoma: a case reportInt J Gynecol Cancer 2006 16(Suppl 1):360-62.10.1136/ijgc-00009577-200602001-0006416515623 [Google Scholar] [CrossRef] [PubMed]
[3]. Shimada S, Tsuzuki T, Kuroda M, Nagasaka T, Hara K, Takahashi E, Nestin expression as a new marker in malignant peripheral nerve sheath tumoursPathology International 2007 57:60-67.10.1111/j.1440-1827.2006.02059.x17300669 [Google Scholar] [CrossRef] [PubMed]
[4]. Alavi S, Arzanian MT, Nilipour Y, Retroperitoneal malignant peripheral nerve sheath tumour replacing an absent kidney in a childCase Rep Oncol Med 2013 2013:62747210.1155/2013/62747224392233 [Google Scholar] [CrossRef] [PubMed]
[5]. Schaefer IM, Fletcher CDM, Recent advances in the diagnosis of soft tissue tumoursPathology 2018 50(1):37-48.10.1016/j.pathol.2017.07.00728950990 [Google Scholar] [CrossRef] [PubMed]
[6]. Taxy JB, Battifora H, Trujillo Y, Dorfman DH, Electron Microscopy in the diagnosis of Malignant SchwanomaCancer 1981 48:1381-91.10.1002/1097-0142(19810915)48:6<1381::AID-CNCR2820480621>3.0.CO;2-6 [Google Scholar] [CrossRef]
[7]. Erlandson RA, Woodruff JM, Peripheral nerve sheath tumours: an electron microscopic study of 43 casesCancer 1982 49(2):273-87.10.1002/1097-0142(19820115)49:2<273::AID-CNCR2820490213>3.0.CO;2-R [Google Scholar] [CrossRef]
[8]. Neilson D, Jones GS, Woodruff JD, Goldberg B, The innervation of the ovaryObstet Gynecol Surg 1970 25:889-912.10.1097/00006254-197010000-00001 [Google Scholar] [CrossRef]
[9]. Nogales FF, Favara BE, Major FJ, Silverberg SG, Immature teratoma of the ovary with a neural component (“solid teratoma”)Hum Pathology 1976 7:625-42.10.1016/S0046-8177(76)80076-7 [Google Scholar] [CrossRef]
[10]. Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Wireman BC, Malignant peripheral nerve sheath tumours state of the science: leveraging clinical and biological insights into effective therapiesSarcoma 2017 2017:742969710.1155/2017/742969728592921 [Google Scholar] [CrossRef] [PubMed]
[11]. Goertz O, Langer S, Uthoff D, Ring A, Stricker I, Tannapfel A, Diagnosis, treatment and survival of 65 patients with malignant peripheral nerve sheath tumoursAnticancer Res 2014 34(2):777-83. [Google Scholar]
[12]. Kar M, Deo SV, Shukla NK, Malik A, DattaGupta S, Mohanti BK, Malignant peripheral nerve sheath tumours (MPNST)--clinicopathological study and treatment outcome of twenty-four casesWorld J Surg Oncol 2006 4:5510.1186/1477-7819-4-5516923196 [Google Scholar] [CrossRef] [PubMed]
[13]. Dover H, Neurofibrosarcoma of the ovary associated with neurofibromatosisCan Med Assoc J 1950 63(5):488-90. [Google Scholar]
[14]. Stone GC, Bell DA, Fuller A, Dickersin GR, Scully RE, Malignant schwannoma of the ovary. Report of a caseCancer 1986 58(7):1575-82.10.1002/1097-0142(19861001)58:7<1575::AID-CNCR2820580732>3.0.CO;2-1 [Google Scholar] [CrossRef]