Nitisinone-Induced Keratopathy in Alkaptonuria Disease: A Case Report and Literature Review
Khalid Mousa Al Zubi1, Mohammed Salem Alsbou2, Mahmoud Hussein Alkhasawneh3, Hani Mosleh Al-Shagahin4
1 Assistant Professor, Special Surgery, Mutah University, Amman, Jordan.
2 Associate Professor, Pharmacology, Mutah University, Amman, Jordan.
3 Assistant Professor, Radiology, Mutah University, Amman, Jordan.
4 Associate Professor, Special Surgery, Mutah University, AlKarak, Amman, Jordan.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Khalid Mousa Al Zubi, Assistant Professor, Special Surgery, Mutah University, P.O.Box 1342, Amman, Jordan.
E-mail: dr_khalid zu@yahoo.com
We report a case of nitisinone-induced keratopathy in a 52-year-old male patient with Alkaptonuria (AKU) disease in Jordan. The patient presented with slight drop of left eye vision for the last four weeks, associated with a foreign body sensation in his left eye. On examination, it was found that his left eye vision was reduced to 6/12 and there were left dendrite-like corneal opacities in the left eye. He was given 10 mg of nitisinone per day, two years before his visual complaint. Three months following discontinuation of nitisinone, his visual acuity and corneal opacities disappeared completely. This is the fifth reported case of nitisinone-induced keratopathy, due to secondary tyrosinaemia in alkaptonuria.
Pseudodendritic keratitis, Richner–Hanhart syndrome, Tyrosinaemia
Case Report
A 52-year-old male patient, known case of alkaptonuria, presenting to the Eye Center with a chief complaint of blurred vision and foreign body sensation in the left eye for the last four weeks from the date of presentation.
The patient had history of severe low back pain, morning stiffness, and pain in his both knees and hips 17 years prior to the present complaint. Clinical examination revealed a bluish-grey pigmentation of both ears. He had been passing dark urine since birth. His urine turned dark-black in colour upon standing for 24 hours. A transient blue colour was observed on performing Ferric chloride (FeCl3) test. This indicated a positive test and suggested the presence of Homogentisic Acid (HGA). Confirmation of diagnosis was done by quantitative measurement of HGA in 24 hour urine sample using Gas Chromatography Mass Spectrometry (GC-MS), which revealed a high level of HGA. The patient had a family history of AKU.
After obtaining an informed consent from the patient, he was started on 10 mg of nitisinone per day, two years prior to his chief complaint, but could not comply with a reduced protein diet while he was on nitisinone treatment. Clinical examination revealed, his best corrected visual acuity of the right eye was 6/6 and that of the left eye was 6/12. The right eye examination showed grey scleral pigmentation and a normal posterior segment. The left eye examination showed grey scleral pigmentation and there were dendrite-like corneal opacities [Table/Fig-1] with a few punctate epithelial erosions, with no other signs of active inflammatory or infective keratitis and left eye posterior segment was normal. The intraocular pressure was 16 mmHg in the right eye and 15 mmHg in the left eye. Examination of the corneal sensation was done and showed bilateral symmetrical and normal sensation.
Slit lamp examination of the left eye plus slit lamp photography showing corneal opacities.
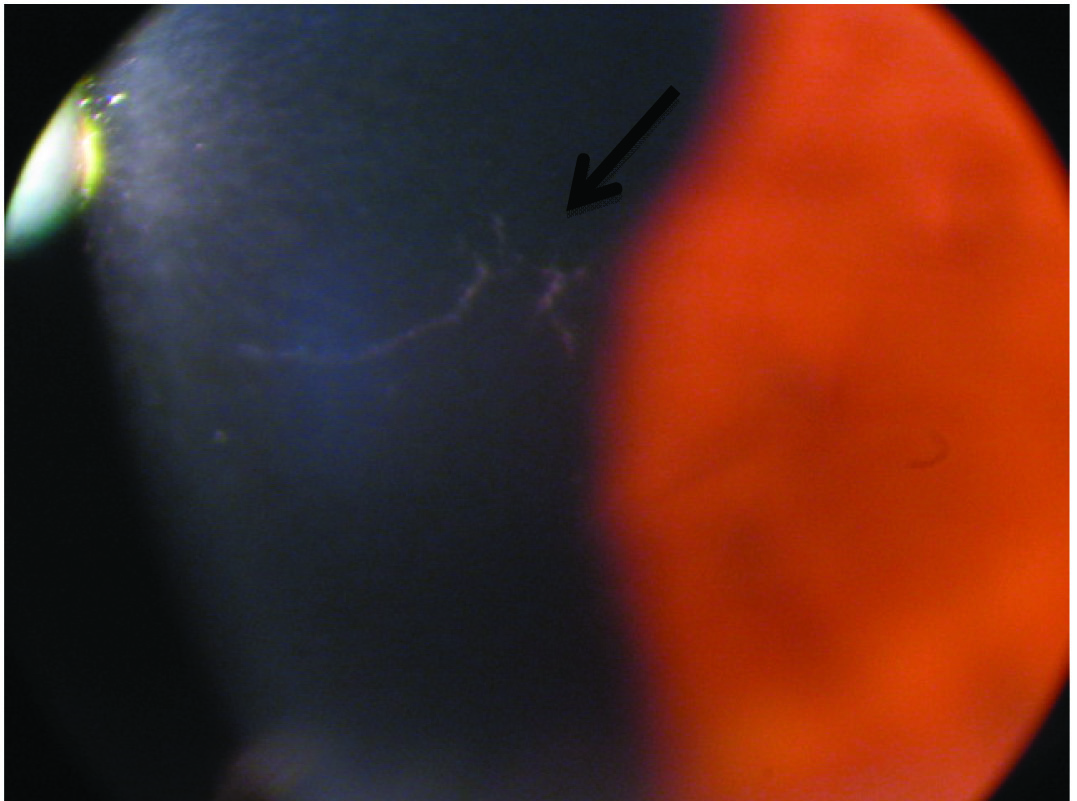
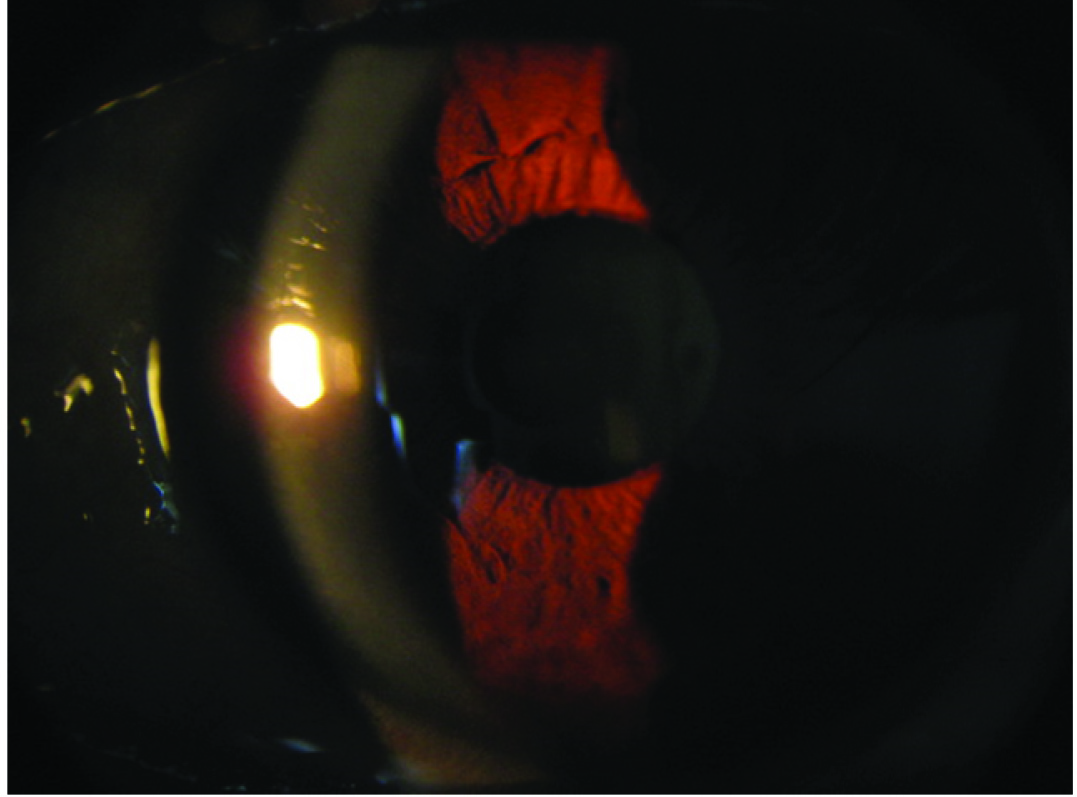
A conjunctival swab was taken and polymerase chain reaction test did not reveal the presence of Herpes simplex virus and Herpes zoster virus infection. A blood sample was also taken and serum tyrosine level was 703.3 μmol/L (reference range 30–90 μmol/L). So, after a thorough discussion with the patient, a decision was taken to stop the nitisinone treatment completely and he was started on sodium hyaluronate eye drop four times daily. Three months later, he was asymptomatic, with the best corrected visual acuity was 6/6 in both eyes, and his corneal opacities were completely resolved [Table/Fig-2]. Serum tyrosine level was also within normal limits at 76.6 μmol/L.
Slit lamp examination of the left eye plus slit lamp photography showing resolved corneal opacities.
Discussion
Alkaptonuria is a very rare inherited metabolic disorder of tyrosine degradation, due to a genetic defect in the enzyme homogentisate dioxygenase, which result in high circulating Homogentisic Acid (HGA) [1,2]. Upon exposure to air, homogentisic acid is oxidized to form a pigment like polymeric material responsible for the black colour of urine. The homogentisic acid is rapidly cleared and excreted by the kidney, but over time it is accumulated and deposited in the connective tissues and converted to a pigment like polymer (ochronosis). This is a chronic process and the external signs of pigment deposition usually start to appear by the fourth decade of life. Ocular signs of alkaptonuria are the black or grey discolouration of the sclera which is the most common finding [3], oil-drop like pigmentation of the limbus which is slightly less common but pathognomonic for alkaptonuria, conjunctival pigmentation and increased conjunctival vessel diameter, severe progressive astigmatism [4,5], and anterior chamber angle hyperpigmentation which may cause secondary glaucoma [3].
The incidence rate of AKU is 1 in 250,000 in most populations. However, high incidence rates have been reported in some countries such as Slovakia (1 in 19,000) [6]. Previous studies have identified 73 cases with AKU in Jordan [7]. Forty cases were diagnosed in one village in the southern region of Jordan, two-third of the patients were young under the age of thirty and nine patients were affected with the disease in one family. The high incidence of AKU in Jordan is due to the high rate of consanguineous marriages [8,9].
Nitisinone {Orfadin; 2(-2-nitro-4 trifluoromethylbenzoyl)-1,3 cyclohexanedione} is an FDA-approved drug for the treatment of hereditary Tyrosinaemia type 1 (HT-1) [10] and it has been widely used at a high dose of 1-2 mg/kg/day in the treatment of this disease [1,11]. It is a new non-licensed treatment for alkaptonuria, which inhibits p-hydroxyphenyl pyruvate dioxygenase, the enzyme leading to the formation of HGA. So, it can drastically reduce urinary excretion of HGA and also reduce HGA in plasma, in individuals with alkaptonuria [12,13].
This mode of action will lead to the increase of the circulating tyrosine (secondary Tyrosinaemia) [1,14]. Tyrosinaemia can be classified into three types: ‘Hereditary Infantile Tyrosinaemia’ (Tyrosinaemia I), ‘Richner-Hanhart Syndrome’ (Tyrosinaemia II) and ‘Tyrosinaemia III’.
The resulting secondary Tyrosinaemia due to the use of nitisinone is more likely to resemble Tyrosinaemia type II [15], where the corneal deposits and skin manifestations are the hallmark of this type.
Tyrosinaemia type II was first described by Richner H and nine years later by Hanhart E as an oculocutaneous syndrome [16,17]. It is a rare disease caused by a deficiency of hepatic Tyrosine Aminotransferase (TAT) with an autosomal recessive mode of inheritance, resulting in elevated plasma and urinary tyrosine concentrations. It is usually characterized by painful palmoplantar hyperkeratotic lesions, bilateral pseudodendritic keratitis, mental retardation, and variable manifestations of central nervous system involvement [15].
Dendrite-like corneal opacities can be seen in Tyrosinaemia type I, Tryosinemia II, Neurotrophic keratopathy, Recurrent Erosion Syndrome, contact lens use, corneal dystrophies and drug toxicity [16-20].
This is the fifth reported case of nitisinone-induced reversible keratopathy due to secondary Tyrosinaemia, the first and second cases were using 2mg/kg/day and 2mg every other day respectively. The third case showed that introducing a low protein diet with low dose of nitisinone was successful in controlling plasma tyrosine levels and the patient remained asymptomatic [21]. However, asymptomatic dendritiform corneal keratopathy has been described with the use of low dose nitisinone in AKU in the fourth case [22]. The significant findings in the five reported cases of nitisinone-induced keratopathy in alkaptonuria disease is summarised in [Table/Fig-3].
Reported cases of Nitisinone-induced Keratopathy in Alkaptonuria disease [1,12,21,22].
| Status of Corneal Findings | Skin Findings | Corneal Findings | Nitisinone Dose | Case Number and Reference |
|---|
| Reversible | None | Branching, sub-epithelial corneal opacities | 2 mg daily | 1. Introne WJ et al., [12] |
| Reversible | itchy urticarial skin rash | Dendritic keratopathy | 2 mg on alternate days | 2. Stewart RM et al., [1] |
| Reversible | None | Corneal crystalline keratopathy | 0.5 mg daily | 3. White A et al., [21] |
| Reversible | None | Asymptomatic dendritiform corneal keratopathy | 2 mg daily | 4. Khedr M et al., [22] |
| Reversible | None | Dendrite-like corneal opacities | 10 mg daily | 5. Present Study |
In all cases, the dendrite-like corneal deposits were completely reversible. Previous studies showed that the plasma tyrosine levels averaged 800 μmol/L, in individuals using Nitisinone in AKU treatment and these tyrosine levels were remarkably well-tolerated [1]. But, in the first reported case using 2 mg of nitisinone per day, the plasma tyrosine level was 200 μmol/L, which is far below the average level in treated individuals suggesting that there are other predisposing factors for the corneal toxicity. In our case the plasma tyrosine level was 703.3 μmol/L on presentation which is around the average levels and it was 76.6 μmol/L three months following discontinuation of the treatment when he was completely asymptomatic with disappearance of his left corneal opacities.
Conclusion
Using nitisinone in the treatment of AKU is still not licensed, but it has a high efficacy in reducing the plasma and urine HGA. This mode of action will result in secondary Tyrosinaemia, especially if there was no restriction of protein intake. Dendrite-like corneal opacities can be seen in individuals using nitisinone, due to the secondary hypertyrosinaemia, but they are usually completely reversible upon discontinuation of the treatment. Thus, we need to be aware of this treatable complication and further research may be required to identify other predisposing factors, and an alternative treatment for AKU.
[1]. Stewart RM, Briggs MC, Jarvis JC, Gallagher JA, Ranganath L, Reversible keratopathy due to hypertyrosinaemia following intermittent low-dose nitisinone in alkaptonuria: a case reportJIMD Reports 2014 17(1)Berlin HeidelbergSpringer:1-6.10.1007/8904_2014_30724997710 [Google Scholar] [CrossRef] [PubMed]
[2]. Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Natural history of alkaptonuriaNew England Journal of Medicine 2002 347(26):2111-21.10.1056/NEJMoa02173612501223 [Google Scholar] [CrossRef] [PubMed]
[3]. Lindner M, Bertelmann T, On the ocular findings in ochronosis: a systematic review of literatureBMC ophthalmology 2014 14(1):1210.1186/1471-2415-14-1224479547 [Google Scholar] [CrossRef] [PubMed]
[4]. Ehongo A, Schrooyen M, Pereleux A, Important bilateral corneal astigmatism in a case of ocular ochronosisBulletin de la Societe belge d’ophtalmologie 2005 (295):17-21. [Google Scholar]
[5]. Cheskes J, Buettner H, Ocular manifestations of alkaptonuric ochronosisArchives of Ophthalmology 2000 118(5):724-25.10.1001/archopht.118.5.72410815172 [Google Scholar] [CrossRef] [PubMed]
[6]. Sršeň Š, Cisárik F, Pásztor L, Harmečko L, Opitz JM, Alkaptonuria in the Trenč ín District of CzechoslovakiaAmerican Journal of Medical Genetics Part A 1978 2(2):159-66.10.1002/ajmg.1320020207263435 [Google Scholar] [CrossRef] [PubMed]
[7]. Alajoulin OA, Alsbou MS, Somayya O, Kalbouneh HM, Spontaneous Achilles tendon rupture in alkaptonuriaSaudi Medical Journal 2015 36(12):148610.15537/smj.2015.12.1283426620992 [Google Scholar] [CrossRef] [PubMed]
[8]. Al-sbou M, Mwafi N, Lubad MA, Identification of forty cases with alkaptonuria in one village in JordanRheumatology International 2012 32(12):3737-40.10.1007/s00296-011-2219-x22147108 [Google Scholar] [CrossRef] [PubMed]
[9]. Al-sbou M, Mwafi N, Nine cases of Alkaptonuria in one family in southern JordanRheumatology international 2012 32(3):621-25.10.1007/s00296-010-1701-121127875 [Google Scholar] [CrossRef] [PubMed]
[10]. Onojafe IF, Adams DR, Simeonov DR, Zhang J, Chan CC, Bernardini IM, Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinismThe Journal of Clinical Investigation 2011 121(10):3914-71.10.1172/JCI5937221968110 [Google Scholar] [CrossRef] [PubMed]
[11]. Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B, Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenaseThe Lancet 1992 340(8823):813-17.10.1016/0140-6736(92)92685-9 [Google Scholar] [CrossRef]
[12]. Introne WJ, Perry MB, Troendle J, Tsilou E, Kayser MA, Suwannarat P, A 3-year randomized therapeutic trial of nitisinone in alkaptonuriaMolecular Genetics and Metabolism 2011 103(4):307-14.10.1016/j.ymgme.2011.04.01621620748 [Google Scholar] [CrossRef] [PubMed]
[13]. Suwannarat P, O’Brien K, Perry MB, Sebring N, Bernardini I, Kaiser-Kupfer MI, Use of nitisinone in patients with alkaptonuriaMetabolism 2005 54(6):719-28.10.1016/j.metabol.2004.12.01715931605 [Google Scholar] [CrossRef] [PubMed]
[14]. Lock EA, Ellis MK, Gaskin P, Robinson M, Auton TR, Provan WM, From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drugJournal of Inherited Metabolic Disease 1998 21(5):498-506.10.1023/A:10054587033639728330 [Google Scholar] [CrossRef] [PubMed]
[15]. Tsai CP, Lin PY, Lee NC, Niu DM, Lee SM, Hsu WM, Corneal lesion as the initial manifestation of Tyrosinaemia type IIJournal of the Chinese Medical Association 2006 69(6):286-88.10.1016/S1726-4901(09)70259-X [Google Scholar] [CrossRef]
[16]. Ahmad S, Teckman JH, Lueder GT, Corneal opacities associated with NTBC treatmentAmerican Journal of Ophthalmology 2002 134:266-68.10.1016/S0002-9394(02)01532-5 [Google Scholar] [CrossRef]
[17]. Iskeleli G, Bilgeç MD, Arici G, Atalay E, Oğreden T, Aydin A, Richner-Hanhart syndrome (Tyrosinaemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesionTurk J Pediatr 2011 53(6):692-94. [Google Scholar]
[18]. Starck T, Kenyon KR, Hanninen LA, Beyer-Machule C, Fabian R, Gorn RA, Clinical and histopathologic studies of two families with lattice corneal dystrophy and familial systemic amyloidosis (Meretoja syndrome)Ophthalmology 1991 98(8):1197-206.10.1016/S0161-6420(91)32153-5 [Google Scholar] [CrossRef]
[19]. Sevim DG, Gumus K, Cavanaghm HD, Corneal pseudodendritic lesions masquerading as herpetic keratitis in a patient with Tyrosinaemia type IEye & Contact Lens 2017 43(3):e7-9.10.1097/ICL.000000000000018726322918 [Google Scholar] [CrossRef] [PubMed]
[20]. Basu PK, Toxic effects of drugs on the corneal epithelium: a reviewJournal of Toxicology: Cutaneous and Ocular Toxicology 1983 2(4-5):205-27.10.3109/15569528309036262 [Google Scholar] [CrossRef]
[21]. White A, Tchan MC, Nitisinone-Induced Keratopathy in Alkaptonuria: A Challenging Diagnosis Despite Clinical SuspicionIn JIMD Reports 2017 Berlin, HeidelbergSpringer:1-3.10.1007/8904_2017_5628879639 [Google Scholar] [CrossRef] [PubMed]
[22]. Khedr M, Judd S, Briggs MC, Hughes AT, Milan AM, Stewart RM, Asymptomatic Corneal Keratopathy Secondary to Hypertyrosinaemia Following Low Dose Nitisinone and a Literature Review of Tyrosine Keratopathy in AlkaptonuriaJIMD Reports 2017 190(1)Berlin, HeidelbergSpringer:1-7.10.1007/8904_2017_6228942493 [Google Scholar] [CrossRef] [PubMed]