CAH comprises a group of autosomal recessively inherited disorders of enzymes involved in steroid biosynthesis. Deficiency of 21-hydroxylase accounts for more than 95% of cases of CAH [1]. This P450 enzyme (CYP21, P450c21) hydroxylates progesterone and 17- hydroxyprogesterone to yield 11-deoxycorticosterone and 11-deoxycortisol. These conversions are required for synthesis of aldosterone and cortisol, respectively. Classic or the severe form of CAH due to 21-hydroxylase deficiency includes the salt losing and simple virilising varieties. It presents during the neonatal period. Both hormones are deficient in the most-severe, “salt-wasting” form of the disease. Less severely affected patients with simple virilising form are able to synthesize adequate amounts of aldosterone but have elevated levels of androgens of adrenal origin. Non classic form of 21 hydroxylase deficiency presents only later in life with features of androgen excess [1].
Based upon neonatal screening studies to detect classic CAH involving 6.5 million neonates worldwide, 21-hydroxylase deficiency is one of the more common inherited disorders occurring in approximately 1 in 15,000 live births [2]. The children with classic congenital adrenal insufficiency apart from presenting with salt losing or shock early in life, also suffer from inappropriate sex assignment at birth, early puberty, short adult stature either due to premature epiphyseal closure or excessive glucocorticoid exposure secondary to treatment, hypertension secondary to treatment, infertility and insulin resistance [3-5].
European Society for Paediatric Endocrinology recommends that surgery should be done in virilised girls with classic CAH at age two to six months. Thus management of CAH requires a multidisciplinary team that includes specialists in paediatric endocrinology, paediatric surgery, urology, psychosocial services and genetics [6].
Only scanty literature is available regarding clinical profile and outcome of neonates presenting with classical CAH. In areas with poor resource settings, routine screening for CAH is not done. The objective of this study was to determine the clinical profile of neonates admitted with classical CAH in our setting, predictors of their outcome and also assess their growth on follow up.
Materials and Methods
We undertook an observational study which had a retrospective part for studying clinical profile, predictors and prospective part for assessing growth of these children on follow up. We conducted this study at a tertiary neonatal care teaching hospital in South India during the months of October 2016 to December 2016. This study was approved by the Institutional Ethics Committee. We reviewed the medical records of neonates diagnosed with classical CAH from September 2006 to September 2016. We took convenient sample size and all the neonates admitted with clinical features, electrolyte abnormalities suggestive of CAH and also with 17-OHP levels higher than the normal gestation based range in the ten years period were included in the study [7].
We collected data from their inpatient records. The parents of these babies were contacted and health status of their children enquired. Assessment of height and weight of these children was carried out during their follow up visit and z scores found by using WHO Charts [8] according to their age. Body weight was measured using a electronic weighing machine with an accuracy of ±10 gm and height measured by a stadiometer with an accuracy of ±1 cm. Short stature was defined as height SDs lesser than -2.0. Blood pressure was checked and drug dosage modified according to 17-OHP levels. A written informed consent was sought from the parents of these children for inclusion in the study.
We used descriptive statistics to describe baseline variables. We compared categorical outcome variables by Chi-square test or Fisher’s-exact test; normally distributed variables by Student’s t-test, variables with skewed distribution by Mann–Whitney U-test. Individual predictors for death were determined by inferential univariate analysis. A p-value< 0.05 was considered statistically significant.
Results
Total admissions to newborn ward in the past 10 years were 38872. Out of this, 40 neonates were diagnosed with classical 21-hydroxylase deficiency. Details regarding their clinical profile, outcome on follow up were collected and growth of these children on follow up was assessed [Table/Fig-1].
Line diagram indicating the flow of the study.
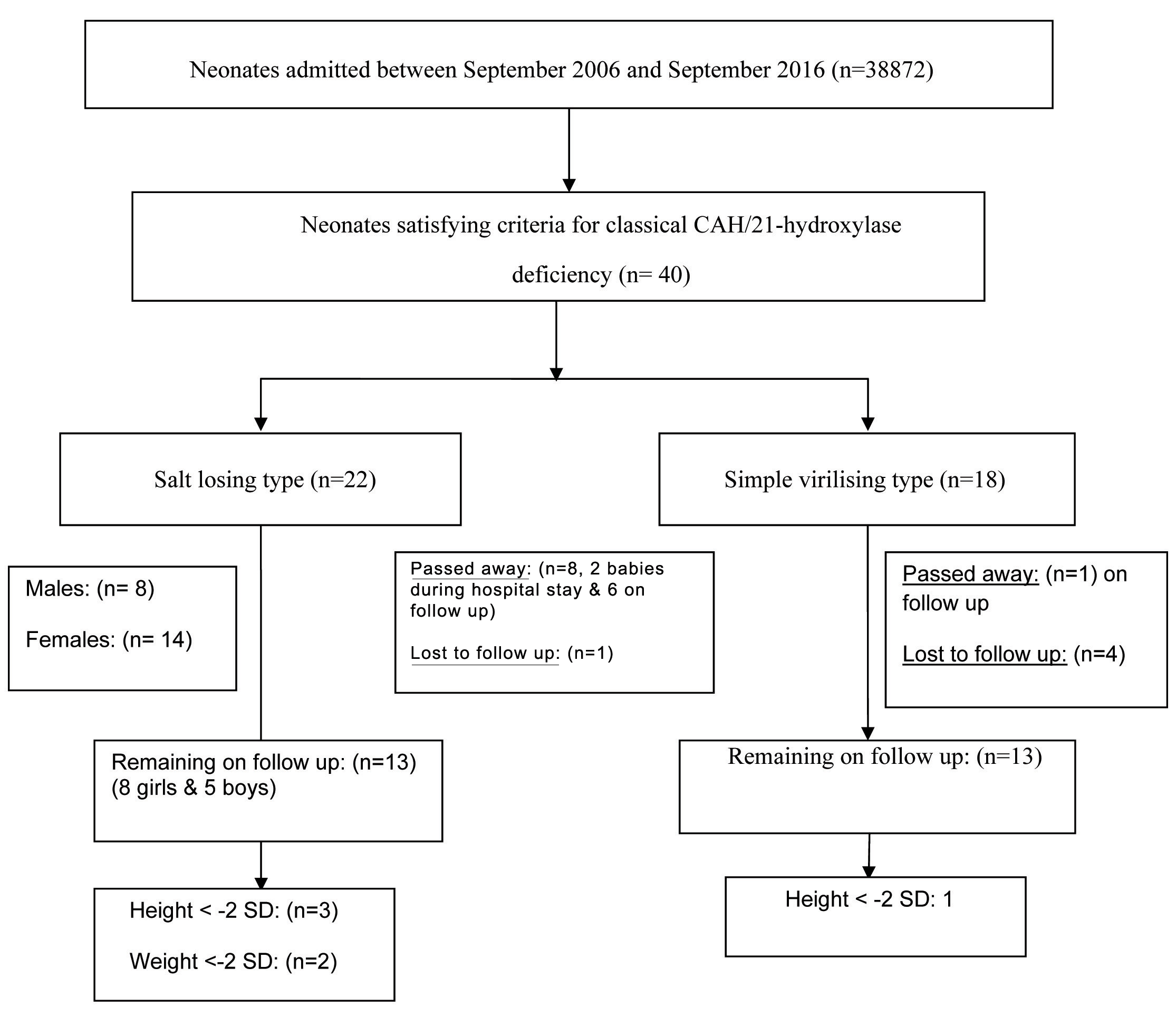
Out of these 40 neonates, 32 were females and 8 were males. Among the female babies, babies (n=20, 62%) were not assigned sex and told to be ambiguous at birth, (n=5, 16%) were assigned female sex and (n=7, 22 %) were assigned male sex at birth. In (n=4, 50%) male babies, CAH was not kept as a possibility at admission.
Among 22 neonates with salt wasting type of CAH, (n=15, 68%) were term babies. Median age at presentation was 10 days with the minimum age being three days and maximum being 27 days. (n=10, 45%) babies had more than 10% weight loss at admission. Lethargy, refusal to be fed and ambigous genitalia were the predominant complaints at the time of admission. Shock was present at admission in (n=11, 50%) babies. (n=17, 77%) babies had hyperpigmentation. (n=5, 23%) had hypoglycaemia. (n=2, 9%) died during hospital stay [Table/Fig-2].
Baseline characteristics of salt losing type of classical CAH.
| Characteristics | Subjects (N=22) |
|---|
| Age at presentation€ | 10 days (3,27) |
| Gestational Age¥ | 37.86±1.2 |
| Birth weight¥ | 2747.73±407.5 |
| Weight loss at admission (%)¥ | 12.3±6.6 |
| Consanguinity | 13(59%) |
| Unexplained sibling death | 2(9%) |
| Referred as suspected CAH | 2(9%) |
| Presenting complaints | Lethargy & Refusal of feeds | 8(36.4%) |
| Lethargy /vomiting/loose stools | 5(22.7%) |
| Appearance of genitalia | Clitoromegaly±Fused labia | 10(45.5%) |
| Hypospadias /Bifid scrotum | 2(9%) |
| Normal male | 8(36.4%) |
| Admission Status | Dehydration | Some | 3(13.6%) |
| Severe | 10(45.5%) |
| Shock | Requiring bolus | 2(9%) |
| Requiring bolus & ionotropes | 9(41%) |
| Requiring intubation | 4(18.2%) |
| Hyperpigmentation | 17(77.3%) |
| Biochemical Abnormality | Hypoglycaemia | 5(22.7%) |
| Hyponatraemia | 22(100%) |
| Hyperkalemia | 20(90.9%) |
| Metabolic acidosis | 12(54.5%) |
| Ultrasound | Mullerian structures | 13(59.1%) |
| Testis | 8(36.3%) |
| No mullerian structures/testis | 1(4.5%) |
| Medication | Median age of starting drugs€ | 12(3,32) |
| Requiring stress dose | 13(59.1%) |
| Need for increase in dose during hospital stay | 5(22.7%) |
| Duration of hospital stay€ | 11.5(3,26) |
| Outcome | Discharged | 20(90.9%) |
| Expired during hospital stay | 2(9.1%) |
| Expired on follow up | 6(27.3%) |
| Age at death(months) | 3(0.2,13) |
| Lost to follow up | 1(4.54%) |
€Median (Min, Max); ¥Mean±SD.
Among 18 babies with simple virilising form of CAH, (n=3, 17%) had more than 10% weight loss at admission. (n=13, 72%) presented with clitoromegaly, fused labial fusion and (n=5, 28%) had severe virilisation with genitalia giving the impression of hypospadias, bifid scrotum. Mullerian structures were visualised in (n=15, 83%) of these babies [Table/Fig-3].
Baseline characteristics of simple virilising type of classical CAH.
| Characteristics | Patients (N=18) |
|---|
| Age at presentation€ | 9(1,21) |
| Gestational age¥ | 37.50±1.97 |
| Birth weight¥ | 2702.78±475.41 |
| Weight loss at admission (%) ¥ | 10.4±4.3 |
| Consanguinity | 10(55.6%) |
| Unexplained sibling death | 2(9%) |
| Referred as suspected CAH | 4(22.22%) |
| Genitalia | Clitoromegaly/fused labia | 13(72.2%) |
| Hypospadias/bifid scrotum | 5(27.8%) |
| Hyperpigmentation | 8(44.44%) |
| Sodium¥ | 138.17±4.06 |
| Potassium¥ | 4.66±0.84 |
| Ultrasound | Mullerian structures | 15(83.3%) |
| No mullerian structures/Testis | 3(16.7%) |
| Mean age of starting drugs€ | 15(3,25) |
| Duration of hospital stay€ | 7.5(3,15) |
| Lost to follow up | 4(22.2%) |
| On follow up | 13(72%) |
€Median (Min, Max);¥ Mean±SD
The babies were asked to come for follow up every month initially, for two months and then every three months for a year. On follow up, six babies with salt losing type died. We tried to find out predictors of death in salt wasting type of CAH by univariate analysis. None among the factors like sex, excessive weight loss, presence of shock, severe dehydration, hyperkalaemia (k>7), hypoglycaemia, increased duration of hospital stay during first admission were statistically significant predictor of death of these children. Compliance to medications among parents of children who survived was significantly higher than those who died [Table/Fig-4].
Predictors of death in salt losing variety of CAH.
| Characteristics at admission & Follow up | Survived(N=13) | Died(N=8) | OR(95%CI) | p-value£ |
|---|
| Male sex | 6(75%) | 2(25%) | 2.57(0.37-17.83) | 0.605 |
| Birth weight <2.5 Kg | 4(66.7%) | 2(33.3%) | 0.75(0.10-5.4) | 0.77 |
| Age at admission< 10 days | 8(66.7%) | 4(33.3%) | 1.60(0.27-9.4) | 0.605 |
| Weight loss at admission>10 % | 5(50%) | 5(50%) | 7.00(0.61-79.8) | 0.117 |
| Presence of shock | 6(60%) | 4(40%) | 1.16(0.20-6.80) | 0.864 |
| Presence of dehydration | 6(50%) | 6(50%) | 3.50(0.50-24.47) | 0.205 |
| Presence of hypoglycaemia | 3(60%) | 2(40%) | 1.11(0.14-8.68) | 0.920 |
| Sodium <125 mEq/l | 8(61.5%) | 5(25%) | 1.04(0.16-6.40) | 0.965 |
| Potassium>7 mEq/l | 5(83.3%) | 1(16.7%) | 0.22(0.02-2.45) | 0.223 |
| Duration of hospital stay >10 days | 8(66.7%) | 4(33.3%) | 1.60(0.27-9.4) | 0.605 |
| Poor compliance to medication | 2(28.6%) | 5(71.4%) | 9.16(1.14-73.23) | 0.037* |
£univariate logistic regression p-value< 0.05 is considered significant
*Fisher’s-exact test
A total of 26 children, 13 salt losing and 13 simple virilising were present on follow up. A cross-sectional assessment of their weight, height was done and charted in WHO z score charts. (n=3, 23%) in salt wasting, (n=1, 7.6%) in simple virilising forms had short stature. (n=2, 15%) in salt wasting had weight z score below - 2 SD [Table/Fig-5].
Follow up of children with classical 21-OH deficiency.
| Characteristics | No (%) |
|---|
| Died during hospital stay | 2 |
| Died after discharge | 7 |
| Lost to follow up (Total) | 5 |
| Follow up subjects (N=26) |
| Children on follow up | Salt losing | 13 |
| Simple virilising | 13 |
| Mean age at follow up(months) | 47±33 |
| Height z score below -2 SD | Total | 4(15.3%) |
| Salt losing | 3 |
| Simple virilising | 1 |
| Weight z score below -2 SD | Total | 2(7.4%) |
| Salt losing | 2 |
| Simple virilising | 0 |
Discussion
Classical 21-hydroxylase deficiency was found in 40 babies out of 38872 admissions in our centre. Our centre is a tertiary care referral unit catering only to the outborn babies. No routine screening programmes are performed for all the admitted babies. CAH is an ideal candidate to be included in newborn screening panel as it satisfies all the Wilson and Jungner’s criteria for screening [9]. Newborns in developed countries throughout the world have been screened through various screening programs for the disease for many decades [10–12].
However, neonatal screening for this defect is not universally followed in developing countries due to the cost, lack of epidemiological data, limited lab facilities and other feasibility issue. Expanded Newborn Screening was started in 2000 at Hyderabad to screen amino acid disorders, congenital hypothyroidism, CAH, G6PD deficiency, biotinidase deficiency, galactosaemia and cystic fibrosis. After testing a total of 18300 babies, the results revealed a high prevalence of congenital hypothyroidism followed by congenital adrenal hyperplasia. The incidence of CAH was found to be 1:2575 from a small sample survey [13]. Both National neonatology forum and other authors recommend newborn screening programme to be introduced in India in a phased manner [9,14]. They strongly recommend routine screening of all newborns for CAH [14].
The major advantage of screening for CAH is the prevention of incorrect assignments of sex and death due to adrenal crisis. However, the interpretation of 17-OHP concentrations in premature or sick newborn infants is difficult. The anxiety generated by false positive results needs to be balanced against the potential advantages of a screening programme [15]. So, neonatal screening program is adapted according to the prevalence of the defect and availability of resources in each setup. Though 10% of these babies with classical 21-hydroxylase deficiency had history of unexplained sibling death in our study, none of them had undergone neonatal screening after birth. Mandatory neonatal screening at least in these babies before they became symptomatic would have given a clue to diagnosis.
Studies on the epidemiology of 21-hydroxylase deficiency from developing countries like Turkey, India and Singapore showed a salt wasting to simple virilising ratio of 1:1.6, 1:1.6 and 1:1.8 respectively [16–18]. In the study by Thilen A et al., in Sweden, 65% had salt wasting form and 35% simple virilising [19]. The number of patients with salt wasting form of 21-hydroxylase deficiency (n= 22, 55%) was slightly higher than those with simple virilising form in our study. Thus, a greater proportion of patients with salt wasting form are being missed in developing countries compared to the developed countries.
Girls with salt wasting form have ambiguous genitalia at birth and are therefore, more likely to be diagnosed, compared to boys who do not have any external abnormality. In our study also, 16 girls (74%) with salt losing form were more than boys. In the study by Bhanji R et al., consanguinity was found in (n=33, 52.3%) cases [20]. As CAH is an autosomal recessive disorder, there are increased chances of this disorder occurring in babies born out of consanguineous marriages. Similarly in our study, 23 patients (57.5%) were born out of consanguineous marriage.
In the study by Bajpai A et al., CAH was not considered in 72% of males with the disease at admission [21]. In our study, the diagnosis of salt wasting form of disease was not considered in (n=3, 50%) male neonates at the time of admission. Hence, in the presence of symptoms like vomiting, excessive dehydration, excessive weight loss, polyuria, hyperpigmentation, biochemical features of adrenal insufficiency and sibling death in an infant, salt wasting form of 21-hydroxylase deficiency should be suspected. After taking sample for 17- OHP level, steroids should be started.
Though simple virilising have less chances of mortality and are picked up earlier due to genital ambiguity, even these cases may suffer adrenal crisis in case of an intercurrent illness [22]. Even in our study one child with simple virilising form passed away following severe lower respiratory tract infection illness on follow up.
Eight girls in our series with 21 hydroxylase deficiency at admission presented with male phenotype and cryptorchidism. This necessitates the need for imaging in males with cryptorchidism to look for mullerian structures. Proper counselling is necessary for compliance. Information regarding nature of the disease, drug dosage, need for increase during any illness and follow up should be given to parents and they should be sensitized. A steroid card or medical alert tag regarding normal steroid dose advised and increase in dose during illness will go a long way in acute stabilisation by health workers nearer to the child’s home before referral to a tertiary centre [23].
Short stature is also a concern in CAH. Failure to achieve target height may be secondary to early puberty and thus early epiphyseal fusion, excess steroid treatment or the salt wasting disease per se [24]. Obesity and hypertension were not found to be significantly higher in this cohort of CAH patients on follow up.
Limitation
The present study has some limitations, part of it being retrospective. Assessment of growth velocity by sequential follow ups would have been better than a cross-sectional one.
Conclusion
In a resource limited setting like ours where routine neonatal screening program is not in practice, a high index of suspicion of CAH in symptomatic babies is needed to prevent mortality. Adequate counselling before discharge and monitoring anthropometry apart from 17-OHP values, blood pressure during follow up are equally important as acute stabilisation.
€Median (Min, Max); ¥Mean±SD.
€Median (Min, Max);¥ Mean±SD
£univariate logistic regression p-value< 0.05 is considered significant
*Fisher’s-exact test