Case Report
A 52-year-old woman, (with no previous medical records in the institution) presented to the Department of Propaedeutic Surgery, with a nonspecific upper abdominal pain, pale stools and dark urine for a week. According to her medical history, Hashimoto thyroiditis and enlargement of the right parotid gland were diagnosed an year ago. Physical examination revealed jaundice and mild tenderness in the upper right quadrant of the abdomen. There was no evidence of ascites, lymphadenopathy or hepatosplenomegaly. The patient was also afebrile and her other vital signs were within normal range.
Biochemical laboratory analysis revealed typical signs of obstructive jaundice (total bilirubin 5.8 mg/dL, direct bilirubin 4.6 mg/dL, alkaline phosphatase 245 U/L, γ-glutamyl transpeptidase 982 U/L), aspartate aminotransferase 284 U/L, alanine aminotransferase 722 U/L, lactate dehydrogenase 438 U/L and borderline levels of serum amylase 105 U/L. Level of C-reactive protein (0.6 mg/L) was normal and serum tumour marker CA 19-9 (267 U/ml) was elevated. Complete blood count was within normal range. Moreover, antibody tests, revealed Antinuclear Antibodies (ANA) positive (+) at dilution 1:160, perinuclear antineutrophil cytoplasmic antibodies (+) antithyroid peroxidase antibodies 280 U/L (0-40).
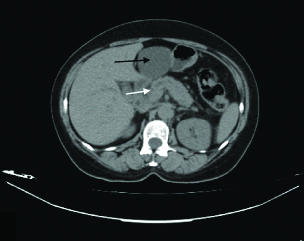
Abdominal ultrasound examination reported a simple liver cyst in the left lobe of the liver, dilatation of intrahepatic biliary ducts, dilatation of Common Bile Duct (CBD; diameter=1 cm). The gallbladder was normal without lithiasis. The patient was then subjected to abdominal computed tomography which confirmed the presence of a liver cyst, with maximum diameter 6.5 cm in the left lobe [Table/Fig-1]. In addition, it showed diffuse enlargement of the head of the pancreas in comparison to the atrophic pancreatic body and tail, slightly dilatated pancreatic duct (3.6 mm) with concomitant CBD dilatation (10 mm), and slight intrahepatic bile duct dilatation.
Computed tomography scan presenting a 6.5 cm liver cyst in the left lobe of the liver (black arrow), diffuse enlargement of the head of the pancreas, a slightly dilatated pancreatic duct (3.6 mm) (white arrow) with concomitant common bile duct dilatation (10 mm), and a slight intrahepatic bile duct dilatation.
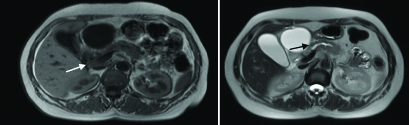
Further diagnostic work up with Magnetic Resonance Imaging (MRI) confirmed the slight dilatation of the pancreatic duct in body and tail of the pancreas till the uncinate process and enlargement of the head of the pancreas [Table/Fig-2]. Hypointense signal on T1 weighted images and relatively hyperintense T2 signal after intravenous contrast infusion was remarkable. No lymph nodes were seen in the retroperioneal, mesenteric region and in the hepatoduodenal ligament. There was also an anatomic variation of the minor pancreatic duct of Santorini, which drained above the ampulla of Vater.
Magnetic resonance imaging presenting a slight dilatation of the pancreatic duct in body and tail of the pancreas (black arrow) and enlargement of the head of the pancreas (white arrow). Hypointense signal on T1 weighted image (left). It is remarkable a T2 hyperintense signal, after intravenous contrast infusion (right).
The diagnostic strategy was to differentiate AIP from pancreatic and biliary cancer. On the other hand; although, there was not any other feature highly suggestive of pancreatic cancer, the case was managed as malignant, unless there was clear evidence of other organ involvement suggestive of AIP.
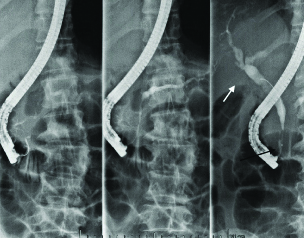
Due to the fact that obstructive jaundice deteriorated, (total bilirubin 9.5 mg/dL, direct bilirubin 6.3 mg/dL), the patient underwent Endoscopic Retrograde Cholangio-Pancreatography (ERCP) [Table/Fig-3]. It revealed oedema in the second part of the duodenum and extreme rigidity of papilla of Vater. A remarkable stenosis was found in the distal part of the common bile duct and dilation of the extra- and intra-hepatic bile ducts. An endoscopic sphincterotomy was performed and a billiary polyethylene stent (10 FR/5 cm) was placed in the common bile duct by an experienced endoscopist.
ERCP presenting stenosis in the distal part of the common bile duct (black arrow) and dilation of the extra- and intra-hepatic bile ducts (white arrow).
Endoscopic Ultrasound (EUS)-Fine Needle Aspiration (FNA) and core biopsy were “gold standard” of the diagnostic process. EUS showed severe hypodensity and heterogeneity of the head of the pancreas with remarkable clear margins (31.8±25.1 mm). The body and tail of the pancreas presented with diffuse heterogeneity, with disseminated hyperdense regions of fibrosis without any other local finding. The pancreatic duct had normal diameter. Regional lymph nodes were revealed, with maximum diameter <1 cm.
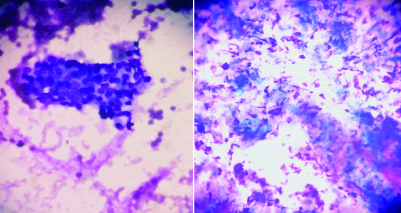
There was no infiltration of the main regional vessels, portal vein, superior mesenteric artery and vein. Cytology revealed clusters of cells with atypia and cellular matrix with inflammatory cells [Table/Fig-4]. Histology revealed dense periductal lymphoplasmocytic infiltration, storiform intralobular fibrosis with secondary atrophy of exocrine acinar component. The IgG4 immunohistochemistry showed dense infiltration of IgG4-positive plasma cells (80 cells per HPF). Fibrosis and scaring involved distal part of common bile duct leading to its obstruction.
Cytology from EUS-FNA (May-Grünwald Giemsa staining, magnification right image 10X, left image 40X) depicting cluster of cells with atypia and cellular matrix with inflammatory cells.
The results of IgG4 in serum were obtained. IgG4 level of 32.1 mg/dL (range normal 8-140 mg/dL) well corresponded with the diagnosis of AIP Type 2.
Therapy
Initial treatment with oral corticosteroids, methylprednisolone 16 mg twice per day for a duration of four weeks was planned. If a response to this regimen was not achieved, surgical intervention would be warranted in the setting of diagnostic confusion regarding pancreaticobiliary malignancy.
One month later, the patient was subjected for another MRI of the abdomen [Table/Fig-5], that showed full remission of the pancreatic and common bile duct dilatation. In addition, diffuse parenchymal enlargement which was the main finding in the first MRI was not observed again. The splenic and portal axis was normal and no regional, paraortic, and mesenteric lymph nodes were found.
Response to steroid therapy. A magnetic resonance imaging of the abdomen revealing full remission of the pancreatic (white arrow) and common bile duct dilatation. Diffuse parenchymal enlargement of the pancreas is not now observed. Note that the liver cyst remained stable (black arrow).
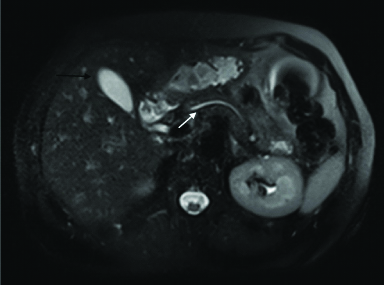
We noted that the only remarkable finding in the subsequent MRI was the presence of the liver cyst which remained stable in size after treatment and the patient was totally asymptomatic.
A gradual taper corticosteroid regimen was followed and the common bile duct stent was removed after three months.
The patient continued low dose of corticosteroids with methylprednisolone for two months, to maintain remission.
Outcome and Follow-up
A follow-up after four and six months after the onset of the disease using MRI and Magnetic Resonance Cholangio-Pancreatography (MRCP), revealed a normal pancreatic gland. Bile and pancreatic ducts were without any significant pathology.
After three years follow-up, patient presented no symptoms; imaging studies and CA 19-9 were within normal range. The liver cyst, however, remained stable in size and totally asymptomatic.
Discussion
Autoimmune pancreatitis is an immune-mediated variant clinical entity of chronic pancreatitis. Yoshida K et al., introduced the term AIP in 1995 to describe an inflammatory pancreatic disease [1]. In 2011, an international panel of experts proposed the International Consensus Diagnostic Criteria (ICDC) for AIP [2,3]. AIP was classified into Type 1 and 2. Type 1 is associated with histological pattern of Lympho-Plasmacytic Sclerosing Pancreatitis (LPSP) which is characterised by IgG4 elevation in serum. Type 2 is known as Idiopathic Duct Centric Pancreatitis (IDCP), expressing Granulocytic Epithelial Lesion (GEL) and is characterised by negative immunoglobulin G4 (IgG4). In this case, IgG4 titres of immunoglobulin were remarkably low, which correlate with the diagnosis of AIP Type 2.
AIP typically presents with painless obstructive jaundice (in about 75% of cases) secondary to entrapment of the intrapancreatic bile duct by the inflammatory process [4] and the diffuse enlargement of the head of the pancreas.
This occurs more often in patients with Type 1 AIP and half of patients with the AIP Type 2. However, patients with Type 2 AIP present more commonly with acute pancreatitis and abdominal pain than patients with Type 1 AIP [5].
Although, genetic and immunogenic factors might play a key role in the development of AIP, its specific pathogenic mechanisms are less well understood [3].
The HISORt criteria [6] that originated from Mayo Clinic were established in 2006. They are based on diagnostic features of AIP which include:
Histology (H)
Imaging (P-Parenchymal; D-Ductal)
Serology (S)
Other Organ Involvement
Response to corticosteroids (Rt)
The [Table/Fig-6], depict major findings of the present case according to HISORt criteria which led to the diagnosis of AIP Type 2.
The diagnosis of Type 2 autoimmune pancreatitis of the present case, according to HISORt criteria.
| Histology | Imaging | Serology | Other organ involvement | Response to corticosteroids |
|---|
| Periductal lymphoplasmocytic infiltration, intralobular fibrosis, dense infiltration of IgG4-positive plasma cells. Fibrosis and scaring of distal part of common bile duct. | CT: Diffuse enlargement of the head of the pancreas, dilated pancreatic duct common bile duct dilatationERCP: Stenosis in the distal part of the common bile duct, dilation of the extra- and intra-hepatic bile ducts. | IgG4 within normal range | Parotid gland involvement | Yes |
Concerning the treatment, AIP is considered to be highly responsive to steroid therapy, making this response a component of the diagnostic criteria [4,7,8]. One commonly used regimen includes treatment with 40 mg of prednisone for four weeks, followed by a taper 5 mg each week for a total of an 11-week course [4]. Patients who relapse, are treated with a second course of corticosteroids. Further, steroid-sparing immunomodulators, such as azathioprine, can be used to maintain remission after relapse [8]. In patients refractory to steroids, azathioprine, mycophenolate mofetil, cyclophosphamide, and rituximab have all been combined with steroid therapy [9].
However, AIP can be a clinical challenge in diagnosis and treatment. This could be attributed to the fact that current diagnostic criteria show some degree of overlap in the findings between AIP and pancreatic cancer, indicated by the similarity in radiologic findings.
In this case, the current patient presented with abdominal pain and jaundice. Radiological imaging examinations, revealed a mass in the head of the pancreas, with dilated bile and pancreatic ducts.
At this point, a potential operation to obtain a surgical specimen for a more accurate histopathology, would be reasonable. However, when we reviewed our case, EUS FNA and core biopsy were ordered to differentiate AIP from pancreatic cancer.
Careful consideration was given to serum IgG4 which were not taken as a special indicator for differential diagnosis between AIP and pancreatic cancer. This is because >20% of all AIP patients have normal IgG4 levels, whereas 7% to 10% of pancreatic cancer patients have elevated IgG4 levels [10].
Synchronous AIP and pancreatic cancer has been reported by Aoyama T et al., who presented a case of pancreatic cancer who underwent pancreaticoduodenectomy. The pathology revealed a well-differentiated adenocarcinoma surrounded by autoimmune pancreatitis, characteristic of lymphoplasmacytic sclerosing pancreatitis [11].
Although, best described in cases of AIP, IgG4-related disease has also been implicated in patients with cholangitis and is now commonly referred to as IgG4-related cholangiopathy. Geary K et al., presented a case of IgG4-related cholangiopathy where they pointed out that it is crucial to differentiate IgG4-related cholangiopathy from its mimickers, such as primary biliary cholangitis, secondary biliary cholangitis, primary sclerosing cholangitis, secondary sclerosing cholangitis, and cholangiocarcinoma [12].
In this regard, AIP could be characterised as a mimicker, masquerading pancreatic cancer. The present patient also had high levels of serum tumour marker CA 19-9, which was indicative of a malignant disease. However, De Marchi G et al., have reported a case with very high serum levels of CA 19-9, higher than 1.000 U/mL found in four patients suffering from AIP [13].
In addition, there are other cases of AIP in the literature that presented as multifocal masses. For instance, Chiang AL et al., have published a case of AIP that was masqueraded as three separate lesions in pancreas [14].
Moreover, AIP is a benign clinical entity that can be effectively treated with corticosteroids and without any interventional treatment. Hence, it must be distinguished from pancreatic cancer to avoid unnecessary surgical resection and prevent delay in the initiation of appropriate treatment. As far as the present case is concerned, interventional treatment with biliary stent was unavoidable, because obstructive jaundice deteriorated the condition of the patient.
There are cases in which AIP was diagnosed after surgical resection. Hoshimoto S et al., reported focal cases of AIP which progressed to more severe grades and exhibited mass formation; although, remaining localised [15]. These patients with AIP were extremely difficult to distinguish from pancreatic cancer. It is also interesting that in this study, a histopathological comparison of mass-forming AIP with the adjacent uninvolved pancreatic tissues was presented but only after surgical treatment.
Finally, high doses of corticosteroids were the initial treatment modality in the present case of AIP Type 2. There are multicentre retrospective studies in the literature that induce spontaneous remission of AIP with steroids [8,14]. Other randomised controlled study, provides evidence of long term maintenance steroid therapy to avoid relapse of AIP [16].
Response to therapy still remains a major criterion of the diagnosis of AIP, according to HISORt criteria.
Conclusion
Autoimmune pancreatitis is a subtype of chronic pancreatitis. Its pathogenic mechanism and combined genetic and immunologic factors need to be clarified. The diagnosis of AIP depends on serology, imaging, histology, other organ involvement and response to steroids. For AIP which mimics pancreatic cancer in clinical and imaging characteristics, further diagnostic workup is necessary to differentiate.
The role of surgery in AIP still remains to be exactly determined in the subgroup of patients with unclear diagnosis or those who fail to respond to corticosteroids. Finally, patients with AIP and endoscopically untreated obstructive jaundice with biliary stenting, might be another subgroup of patients who need surgical biliary drainage. Future similar studies are warranted to understand this finding in depth.
[1]. Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N, Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitisDig Dis Sci 1995 40:1561-68.10.1007/BF022852097628283 [Google Scholar] [CrossRef] [PubMed]
[2]. Shimosegawa T, Chari ST, Frulloni L, Kamisawa T, Kawa S, Mino-Kenudson M, International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the international association of pancreatologyPancreas 2011 40:352-58.10.1097/MPA.0b013e3182142fd221412117 [Google Scholar] [CrossRef] [PubMed]
[3]. Cai O, Tan S, From pathogenesis, clinical manifestation, and diagnosis to treatment: an overview on autoimmune pancreatitisGastroenterol Res Pract 2017 2017:324645910.1155/2017/324645928197205 [Google Scholar] [CrossRef] [PubMed]
[4]. Hoffmanova I, Gurlich R, Janik V, Szabo A, Vernerova Z, Dilemmas in autoimmune pancreatitis. Surgical resection or not?Bratisl Lek Listy 2016 117:463-67.10.4149/BLL_2016_09027546699 [Google Scholar] [CrossRef] [PubMed]
[5]. Kamisawa T, Chari ST, Giday SA, Kim M, Chung JB, Lee KT, Clinical profile of autoimmune pancreatitis and its histological subtypes: an international multicenter surveyPancreas 2011 40:809-14.10.1097/MPA.0b013e3182258a1521747310 [Google Scholar] [CrossRef] [PubMed]
[6]. Chari ST, Smyrk TC, Levy MJ, Topazian MD, Takahashi N, Zhang L, Diagnosis of autoimmune pancreatitis: the Mayo Clinic experienceClin Gastroenterol Hepatol 2006 4:1010-16.10.1016/j.cgh.2006.05.01716843735 [Google Scholar] [CrossRef] [PubMed]
[7]. Majumder S, Takahashi N, Chari ST, Autoimmune pancreatitisDig Dis Sci 2017 62(7):1762-69.10.1007/s10620-017-4541-y28365915 [Google Scholar] [CrossRef] [PubMed]
[8]. Pannala R, Chari ST, Corticosteroid treatment for autoimmune pancreatitisGut 2009 58:1438-39.10.1136/gut.2009.18329319834112 [Google Scholar] [CrossRef] [PubMed]
[9]. Sandanayake NS, Church NI, Chapman MH, Johnson GJ, Dhar DK, Amin Z, Presentation and management of post-treatment relapse in autoimmune pancreatitis/immunoglobulin G4-associated cholangitisClin Gastroenterol Hepatol 2009 7:1089-96.10.1016/j.cgh.2009.03.02119345283 [Google Scholar] [CrossRef] [PubMed]
[10]. Cao Z, Tian R, Zhang T, Zhao Y, Localized autoimmune pancreatitis: report of a case clinically mimicking pancreatic cancer and a literature reviewMedicine (Baltimore) 2015 94(42):e165610.1097/MD.000000000000165626496272 [Google Scholar] [CrossRef] [PubMed]
[11]. Aoyama T, Murakawa M, Atsumi Y, Kazama K, Shiozawa M, Kobayashi S, A case of pancreatic cancer associated with autoimmune pancreatitisGan To Kagaku Ryoho 2016 43:1289-91. [Google Scholar]
[12]. Geary K, Yazici C, Seibold A, Guzman G, IgG4-related cholangiopathy and its mimickers: a case report and review highlighting the importance of early diagnosisInt J Surg Pathol 2017 1:106689691773090210.14309/00000434-201710001-01316 [Google Scholar] [CrossRef]
[13]. De Marchi G, Paiella S, Luchini C, Capelli P, Bassi C, Frulloni L, Very high serum levels of CA 19-9 in autoimmune pancreatitis: report of four cases and brief review of literatureJ Dig Dis 2016 17:697-702.10.1111/1751-2980.1240327579898 [Google Scholar] [CrossRef] [PubMed]
[14]. Chiang AL, Hornick JL, Sahni VA, Clancy TE, Ryou M, Autoimmune pancreatitis presenting as multifocal masses, diagnosed on ampullary biopsyPancreas 2016 45:e25-e27.10.1097/MPA.000000000000058127295535 [Google Scholar] [CrossRef] [PubMed]
[15]. Hoshimoto S, Aiura K, Tanaka M, Shito M, Kakefuda T, Sugiura H, Mass-forming type 1 autoimmune pancreatitis mimicking pancreatic cancerJ Dig Dis 2016 17:202-09.10.1111/1751-2980.1231627121246 [Google Scholar] [CrossRef] [PubMed]
[16]. Masamune A, Nishimori I, Kikuta K, Tsuji I, Mizuno N, Iiyama T, Research committee of intractable pancreas diseases in Japan. Randomised controlled trial of long-term maintenance corticosteroid therapy in patients with autoimmune pancreatitisGut 2017 66:487-94.10.1136/gutjnl-2016-31204927543430 [Google Scholar] [CrossRef] [PubMed]