Case Report
Case-1
A 16-year-old female patient presented to the emergency department with a chief complaint of severe chest pain since three days which was precordial in location and increased on inspiration and with change of posture. There was no history of fever, headache, cough, sputum production, shortness of breath, loss of appetite, decreased urine output, haematuria and weight loss. There was no history of joints pain, skin rash, oral ulcer, photosensitivity or Raynaud’s phenomenon. Patient was already on category I Anti Tubercular Therapy (ATT) for one month for episcleritis of left eye. There was no other significant medical or surgical illness in the past. Menstrual history was also normal.
On examination, her vitals were stable and she was afebrile. There was no pallor, cyanosis, clubbing, icterus, lymphadenopathy or pedaloedema. On cardiovascular examination, heart sounds were normal but a pericardial rub was heard. Respiratory, abdominal and Central Nervous System (CNS) examination did not reveal any significant abnormality.
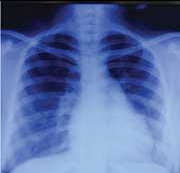
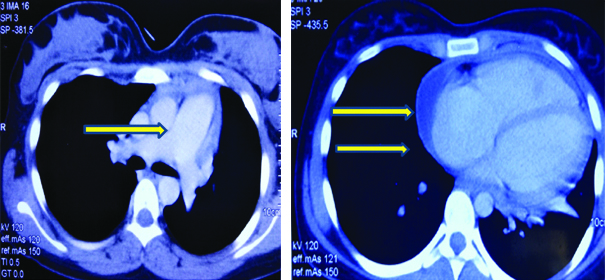
On investigations, haemoglobin was 8.8 gm/dL with a normal total leucocyte count (6,370/mm3) and platelet count of 2.65 lacs/mm3. Erythrocyte Sedimentation Rate (ESR) was raised to 89 mm/hour. Arterial Blood Gas (ABG) analysis was normal. Mantoux test was negative. Serum biochemistry including KFT, Liver function tests, serum calcium, phosphorus and Alkaline Phosphatase test (ALP) and urine routine microscopy with urine for active sediments were normal. Electrocardiogram (ECG) showed T wave inversions in leads V1-V6 and chest X-ray [Table/Fig-1]was suggestive of cardiomegaly. Based on above findings, 2D-echocardiography was done which revealed mild pericardial effusion and moderate pulmonary artery hypertension. A Contrast Enhanced Computed Tomography (CECT) chest revealed cardiomegaly with enlarged right sided chambers with prominent main pulmonary arterial trunk suggestive of pulmonary arterial hypertension with pericardial effusion [Table/Fig-2]. The patient was put on supportive treatment of antipyretics and analgesics till further investigations.
Chest X-ray (posteroanterior view) of the patient suggestive of cardiomegaly.
A contrast enhanced computed tomography chest of the patient is suggestive of cardiomegaly with enlarged right sided chambers with prominent main pulmonary arterial trunk suggestive of pulmonary arterial hypertension with pericardial effusion (yellow arrows).
During the course of hospital stay after four days, the patient suddenly developed pain and blurring of vision of the right eye. She immediately underwent a slit lamp examination which revealed 3+ anterior chamber cells, diffuse, Keratic Precipitates (KPs), vitrits and panscleritis.
In view of the above clinical presentation of acute pericarditis, bilateral panophthalmitis and pulmonary artery hypertension, patient was screened for an autoimmune pathology. Serum Anti Nuclear Antibody (ANA), dsDNA, HLA B-27, rheumatoid factor, Anti-Cyclic Citrullinated Peptide (Anti-CCP), p-ANCA (ELISA), c-ANCA (ELISA), HbsAg, anti-HCV and HIV serology were negative. Serum complement levels of C3 and C4 were normal. However, ANCA by Indirect Immunoflorescence (IIF) was positive for cytoplasmic pattern (titer 1:120). Hence, a final diagnosis of GPA presenting as an isolated necrotising scleritis was made. Patient was given six cycles of cyclophosphamide pulse, along with oral steroids and supportive treatment. Thereafter, she was kept on maintenance therapy of azathioprine (200 mg till date). Currently, the patient is on regular follow-up with no relapse or systemic complications.
Case-2
A 40-year-old female presented to the emergency with complaints of sudden onset breathlessness, low grade fever and non productive cough since four to five days. There was no history of chest pain, palpitations, anorexia, weight loss and haemoptysis. There was no history of joints pain, oral ulcer, photosensitivity, decrease in urine output or any bleeding manifestations. She had a history of tingling and numbness in bilateral feet and hands since the last four months. She also had a bad obstetric history (G8P3L3A5) but normal menstrual cycles. There was no personal history of diabetes and hypertension.
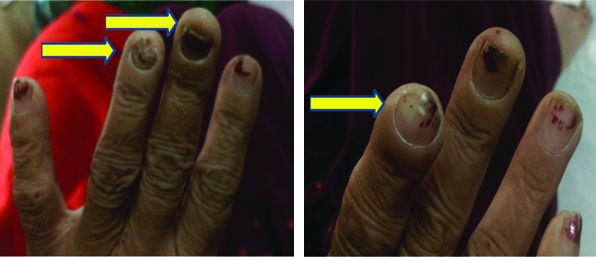
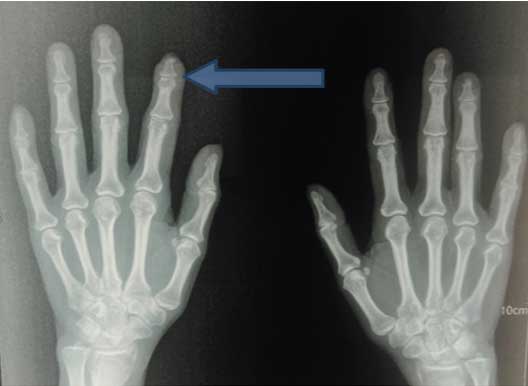
On examination, her vitals were stable and was febrile to touch. There was marked pallor but no cyanosis, icterus, lymphadenopathy or pedaloedema. On local examination, the patient had nail deformities with acro-osteolysis [Table/Fig-3]. She also had a petechial rash which was non tender and palpable over bilateral lower limbs. Auscultation of the chest revealed fine crepitations over bilateral lung fields. Rest of the cardiovascular, abdominal and CNS examination were normal.
Nail deformities with acro-osteolysis (yellow arrows).
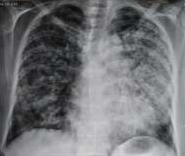
On further investigation, haemoglobin was 4.6 gm/dL with a normal total leucocyte (5,670/mm3) and platelet count (3.45 lacs/mm3). ESR was raised to 110 mm/hour. The ABG analysis was suggestive of type 1 respiratory failure with a SpO2 of 76.8 mmHg. The KFT were deranged with blood urea of 256 mg/dL (normal 20-40) and serum creatinine of 5.6 mg/dL (normal 0.5-1.2) with normal sodium and potassium levels. Rest of the serum biochemistry including LFT, serum calcium, phosphorus and ALP were normal. Urine analysis revealed 30-35 Red Blood Cells per High Power Field (RBC/HPF) with 24 hour urine protein of 2.570 gm/day. It further revealed active sediments with significant proteinuria (+3) with 40% dysmorphic RBC (20-25/hpf). Chest X-ray was suggestive of bilateral diffuse homogenous, interlobar and fluffy opacities in the lung fields [Table/Fig-4].
Chest X-ray (posteroanterior view) view suggestive of bilateral diffuse homogenous, interlobar and fluffy opacities in the lung fields.
Based on the above atypical clinical scenario of sudden onset breathlessness, deranged KFT, active urine sediments with proteinuria and marked pallor without any evidence of overt bleeding and other findings on history and examination, a clinical diagnosis of DAH due to a vasculitic aetiology PRS was made. Immediately, work up of autoimmune pathology was sent and meanwhile, the patient was started on cyclophosphamide and pulse steroids.
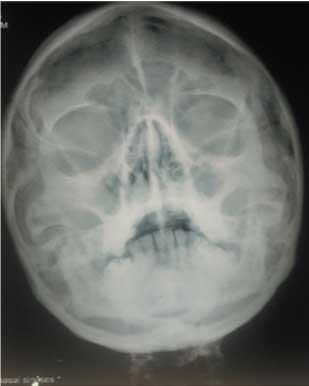
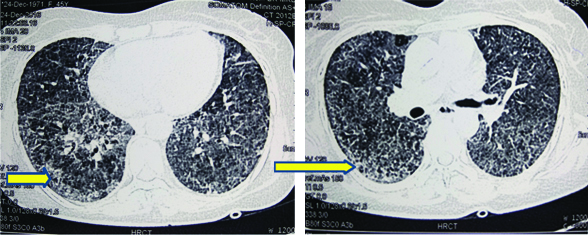
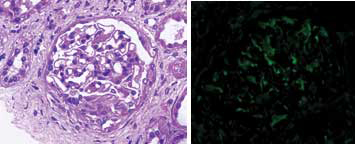
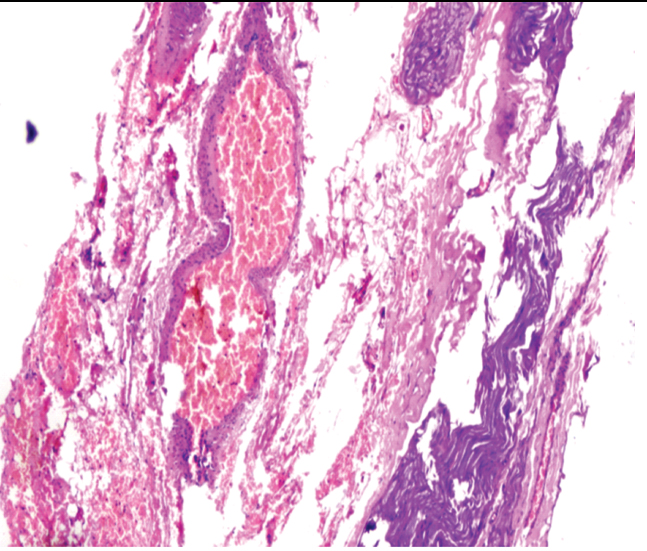
Meanwhile, C-Reactive Protein (CRP) came out to be positive (43.8 mg/dL), X-ray Paranasal Sinus (PNS) was suggestive of bilateral maxillary sinusitis and X-ray bilateral hands with wrists revealed multiple erosions with acro-osteolysis of right second distal phalanx [Table/Fig-5,6]. Serology for HIV, hepatitis B and C was negative. Ultrasound of the abdomen and fundus was normal. ANA was +1 positive, dsDNA was negative, c-ANCA (ELISA) was positive (156 U/mL) along with antiproteinase-3 (PR-3) and p-ANCA was negative. High Resolution Computed Tomography (HRCT) chest was suggestive of diffuse interstitial septal thickening and ground glass opacities with crazy paving suggestive of alveolar haemorrhage [Table/Fig-7]. Renal biopsy was done in view of active sediments in the urine examination which revealed presence of crescentric (>50%), pauci-immune glomerulonephritis [Table/Fig-8]. Nerve Conduction Velocity (NCV) tests revealed predominant sensorimotor neuropathy in bilateral lower limbs. Sural nerve biopsy was done which revealed medial sclerosis of small blood vessels with no evidence of vasculitis. Transbronchial lung biopsy was done which suggestive of DAH [Table/Fig-9]. Repeat chest X-ray was done after three days that showed marked resolution of the bilateral fluffy opacities [Table/Fig-10].
X-ray paranasal sinus view suggestive of bilateral maxillary sinusitis.
X-ray of bilateral hands with wrists showing acro-osteolysis of right second distal phalanx (blue arrow) with multiple erosions.
HRCT chest suggestive of diffuse interstitial septal thickening and ground glass opacities with crazy paving suggestive of alveolar haemorrhage (yellow arrows).
Renal biopsy with immunohistochemistry suggestive of crescentric (>50%), pauci-immune glomerulonephritis (rapidly progressive glomerulonephritis, at 40X magnification).
Transbronchial lung biopsy of the patient is suggestive of multiple areas of haemorrhage scattered in the given tissue with occasional haemosiderin laden macrophages suggestive of diffuse alveolar haemorrhage (H&E stain 10X magnification).
Repeat chest X-ray (posteroanterior view) view of the patient three days after treatment showing marked resolution of the diffuse homogenous opacities as compared to [Table/Fig-4].
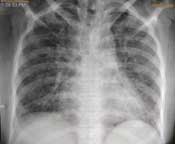
Hence, the final diagnosis of GPA presenting as PRS with DAH was made. The patient responded dramatically to cyclophosphamide (six cycles) and steroids with resolution of symptoms after three days. She was discharged after 2.5 months of hospital stay with a normal chest X-ray and KFT and is currently on follow up on mycophenolate mofetil with no other systemic complications so far.
Discussion
The histopathological hallmarks of GPA are the presence of necrotising vasculitis of small sized arteries and veins along with non caseating granuloma formation, with pathognomic triad of presentation in only 25-30% of the patients, that being the involvement of upper and lower respiratory tracts along with glomerulonephritis [1]. Though, the most common presentation is of the upper respiratory tract in the form of sinusitis and nasal disease (73% cases), isolated involvement of the eyes and PRS is quite rare that too without any chronic clinical course and systemic involvement at presentation and in the follow up period. Up to 46-50% of patients with GPA have ophthalmic manifestations and incidence of DAH has been reported as between 7-45%. If not diagnosed early, both have a high mortality rate of around 50-60% [1,2].
Review of studies on GPA by Filocamo G et al., revealed that around 8-15% cases of GPA occur in the age group of less than 19 years [3]. 158 patients with GPA were analysed by Hoffman GS et al., in which eye disease was present in 52% (with 10% having scleral manifestation of the disease), in which 8% developed blindness [4]. In another review by Bullen CL et al., 29% had ocular disease, out of 140 GPA cases studies retrospectively. The most common was orbit (15%), followed by scleral (7%), episcleral (3.5%) and corneal (8%) [5]. Harper SL et al., found that in a study of 47 patients with GPA, systemic disease followed by ocular disease was present in 57.4%, whereas only 6.3% presented first with eye disease, followed by systemic disease [6].
A Pubmed search using mesh terms such as “GPA, ocular”, “wegeners, ocular” and “wegeners, scleritis” was made and a list has been complied on ocular involvement as a first presentation of GPA [Table/Fig-11].
Ocular involvement as the first presentation of GPA.
| S. no. | Studies | Age/ Sex | Ophthalmological involvement | Treatment given | Outcome |
|---|
| 1. | Lu CW et al. [2] | 40 yr/F | Peripheral ulcerative keratitis | Cryotherapy and oral prednisone and cyclophosphamide | Survived |
| 2. | Siddiqui S et al. [7] | 38 yr/F | Orbital apex syndrome | Steroids and antibiotics | Survived |
| 3 | Sonawale A et al. [8] | 44 yr/F | Central artery retinal occlusion | Steroids, cyclophosphamide | Survived |
| 4. | Franco CM et al. [9] | 60 yr/F | Keratoscleritis (right eye) | Steroids, cyclophosphamide | Survived |
| 5. | Fenech M and Fenech T [10] | 22 yr/F | Bilateral orbital psuedo tumour | Steroids, methotrexate | Survived |
| 6 | Majai G et al. [11] | 38 yr/F | Orbital psuedo tumour | Steroids, cyclophosphamide, rituximab | Survived |
| 7. | Masuda T et al. [12] | 83 yr/F | Choroidal tumour | Steroids, cyclophosphamide, azathioprine | Survived |
| 8. | Dunne K et al. [13] | 84 yr/M | Steroids, methotrexate | Systemic prednisone, cyclophosphamide and prophylactic cotrimoxazole | Survived |
| 9. | Present case report | 16 yr/F | Ocular cicatricial scarring and symblepharon formation | Systemic prednisone, cyclophosphamide pulse (six cycles) with maintenance azathioprine | Survived |
yr: Years; F: Female; M: Male
Similarly, although, the lung is the target organ most commonly affected in GPA, DAH is a rare manifestation of the disease, occurring only in 5-10% of patients and carries an extremely high mortality rate of around 60-70% [14]. More than half of the affected patients have renal involvement in which 70-80% develops pauci-immune necrotising crescentic glomerulonephritis. The most common organ involved is the lung (50-90%), which includes PRS (defined as a combination of DAH and glomerulonephritis) as a rare complication which has a high mortality rate [15]. Compilation of case reports using mesh words on Pubmed “Pulmonary renal syndrome, GPA” and “DAH, GPA” of PRS as a first presentation of GPA is given in [Table/Fig-12].
Pulmonary renal syndrome as a first presentation of GPA.
| S. no. | Studies | Age/ Sex | Treatment given | Plasmapharesis | Outcome |
|---|
| 1. | Chachar AZ et al, [16] | 4o yr/F | Pulse steroids, cyclophosphamide | No | Survived |
| 2. | Kundu S et al, [17] | 42 yr/M | Pulse steroids, cyclophosphamide | Yes | Survived |
| 3. | Al bshabshe AA et al, [18] | 21 yr/F | Pulse steroids, cyclophosphamide, haemodialysis | Yes | Survived |
| 4. | Present case report | 40 yr/F | Pulse steroids, cyclophosphamide, maintenance therapy with MMF (Mycophenolate Mofetil) | No | Survived |
yr: Years; F: Female; M: Male
The diagnosis of GPA requires positive ANCA, mostly cANCA for PR3, which when positive in combination has a 100% positive predictive value for GPA [19]. However, when the illness is “limited” to the orbit or an isolated organ involvement is present, ANCA may be negative in up to 50% of cases [19]. In that scenario, a biopsy of affected organ may be required to determine the diagnosis. Even in present first case, cANCA was negative with ELISA but positive on IIF (Indirect immunoflorence).
The treatment of GPA with corticosteroids and immunosuppressive agents has revolutionised the management of such patients, but morbidity and mortality are still high. In the first case report, cyclophosphamide led to improvement in the ocular manifestations of the patient. Here, a renal biopsy was not done because the KFT, urine microscopy with active sediments was normal. Till now, the patient does not have any systemic involvement. In the second case report, pulse therapy with cyclophosphamide and steroids dramatically improved the patient’s condition with no need for rituximab or plamapheresis. Both the patients are currently on follow-up on maintenance therapy.
Conclusion
The GPA is a challenging, multifaceted disease that requires prompt diagnosis and institution of immunosuppressive therapy. It has a high mortality rate if diagnosed late. Proper follow-up during treatment is needed due to the high rate of relapse. The presence of isolated organ involvement, like evident in the present case report, that is necrotising scleritis and PRS, with no response to standard antibiotic treatment should lead to a suspicion of an underlying vasculitic pathology. Necrotising scleritis often leads to vision loss and blindness if not adequately treated, which is a source of significant morbidity to the patient. Timely institution of immunosuppressive therapy with other supportive case results in dramatic improvement. Follow-up is essential to look out for further development of the systemic illness, which fortunately did not develop in both our patients. Furthermore, early treatment with appropriate medication culminates in improved survival for the individual.
yr: Years; F: Female; M: Male
yr: Years; F: Female; M: Male
[1]. Savage CO, Harper L, Adu D, Primary systemic vasculitisThe Lancet 1997 349(9051):553-58.10.1016/S0140-6736(97)80118-3 [Google Scholar] [CrossRef]
[2]. Lu CW, Liu XF, Luan Y, Lu CB, Zhou DD, Guo LM, A case report of the orbit, ocular association and the lung in granulomatosis with polyangiitis: a diagnostic challengeExp Ther Med 2017 13(6):3337-40.10.3892/etm.2017.440828587410 [Google Scholar] [CrossRef] [PubMed]
[3]. Filocamo G, Torreggiani S, Agostoni C, Esposito S, Lung involvement in childhood onset granulomatosis with polyangiitisPediatric Rheumatology 2017 15(1):2810.1186/s12969-017-0150-828410589 [Google Scholar] [CrossRef] [PubMed]
[4]. Hoffman GD, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, Wegener granulomatosis: an analysis of 158 patientsAnn Intern Med 1992 116(6):488-98.10.7326/0003-4819-116-6-4881739240 [Google Scholar] [CrossRef] [PubMed]
[5]. Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA, Ocular complications of wegener’s granulomatosisOphthalmology 1983 90(3):279-90.10.1016/S0161-6420(83)34574-7 [Google Scholar] [CrossRef]
[6]. Harper SL, Letko ER, Samson CM, Zafirakis P, Sangwan V, Nguyen Q, wegener’s granulomatosis: the relationship between ocular and systemic diseaseJ Rheumatol 2001 28(5):1025-32. [Google Scholar]
[7]. Siddiqui S, Kinshuck AJ, Srinivasan VR, Orbital apex syndrome secondary to granulomatosis with polyangiitisBMJ Case Reports 2013 2013:bcr201300951910.1136/bcr-2013-00951924311409 [Google Scholar] [CrossRef] [PubMed]
[8]. Sonawale A, Rajadhyaksha A, Rajadhyaksha S, Oculo-otological manifestations in a case of granulomatosis with polyangiitisJ Assoc Physicians of India 2017 65(4):82 [Google Scholar]
[9]. Franco CM, Oliveira GM, Fidelix TS, Vieira LA, Trevisani VF, Nodular scleritis and granulomatous polyangiitis (wegener) mimicking tuberculosisRevista Brasileira de Oftalmologia 2015 74(2):106-09.10.5935/0034-7280.20150024 [Google Scholar] [CrossRef]
[10]. Fenech M, Fenech T, Orbital pseudo tumour masquerading as wegener’s granulomatosisMalta Medical Journal 2015 27(3):44-48. [Google Scholar]
[11]. Majai G, Tarjan P, Dezso B, Zeher M, Orbital pseudotumor-a rare variant of granulomatosis with polyangiitisAustin J Clin Immunol 2015 2(1):1024 [Google Scholar]
[12]. Masuda T, Izumi Y, Takeshita H, Kawahara C, Tsuji Y, Kurohama H, Granulomatosis with polyangiitis presenting as a choroidal tumorCase Reports Rheumatol 2015 :27182310.1155/2015/27182325949841 [Google Scholar] [CrossRef] [PubMed]
[13]. Dunne K, Khalid M, A case of granulomatosis with polyangiitis presenting with significant ocular cicatricial scarring and symblepharon formationAme J Ophthalmol Case Rep 2016 2:11-13.10.1016/j.ajoc.2016.06.01029503914 [Google Scholar] [CrossRef] [PubMed]
[14]. Powers B, Uppalapati A, Gogineni S, Jamkhana ZA, Rituximab-A drug with many facets and cures: a treatment for acute refractory hypoxemic respiratory failure secondary to severe granulomatosis with polyangiitisCase Rep Crit Care 2013 :12313429503914 24829812 [Google Scholar] [CrossRef] [PubMed]
[15]. Syed R, Rehman A, Valecha G, El-Sayegh S, Pauci-immune crescentic glomerulonephritis: an ANCA-associated vasculitisBioMed Res Int 2015 :40282610.1155/2015/40282626688808 [Google Scholar] [CrossRef] [PubMed]
[16]. Chachar AZ, Sabir O, Haider I, Tanvir I, Rafique K, Tarif N, Pulmonary renal syndrome in a patient with vasculitis: case report and review of literaturePak J Med Sci 2015 31(6):1545-48.10.12669/pjms.316.839126870133 [Google Scholar] [CrossRef] [PubMed]
[17]. Kundu S, Misra S, Halder RK, Roychowdhury A, Pulmonary renal syndrome in a case of Wegener’s granulomatosisIndian J Chest Dis Allied Sci 2013 55(1):49-52. [Google Scholar]
[18]. Al bshabshe AA, Al-Khalidy H, Omer HA, Al-Amri DH, Hamad A, Abdulwahed SR, Pulmonary renal syndrome associated with wegener’s granulomatosis: a case report and review of literatureClinical Exp Nephrol 2010 14(1):80-84.10.1007/s10157-009-0223-119730971 [Google Scholar] [CrossRef] [PubMed]
[19]. Radice A, Bianchi L, Maggiore U, Vaglio A, Sinico RA, Comparison of PR3-ANCA specific assay performance for the diagnosis of granulomatosis with polyangiitis (Wegener’s)Clinical Chemi Lab Med 2013 51(11):2141-49.10.1515/cclm-2013-030823798618 [Google Scholar] [CrossRef] [PubMed]