Case Report
We report a case of a 56-year-old male non smoker, known hypertensive with history of right lower limb deep vein thrombosis. He was on oral anticoagulation, namely ‘Warfarin’, for three years and stopped since six months. He presented with complaints of neck pain radiating to shoulder, generalised headache, vomiting, and blurring of vision. His vitals were stable and there was no neurological deficit. Abdominal examination showed no splenomegaly.
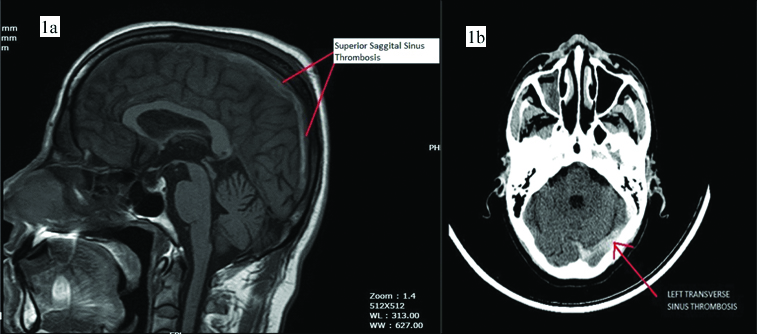
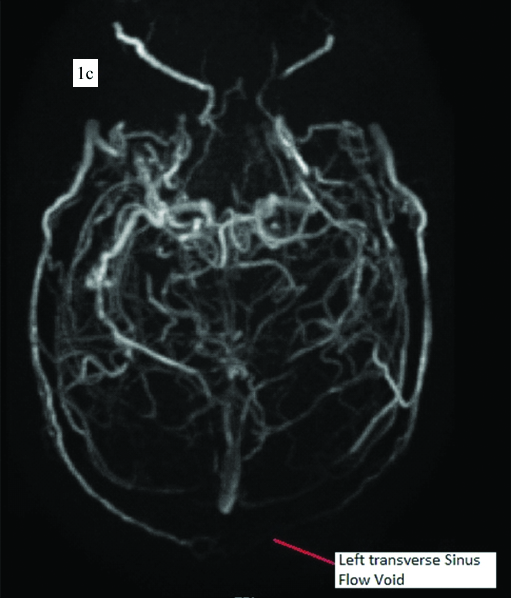
Electrocardiogram (ECG) showed ‘Q’ waves with ‘T’ inversions in inferior leads s/o evolved inferior wall MI. Echocardiogram showed inferior wall hypokinesia, normal biventricular systolic function with trivial mitral regurgitation, no evidence of patent foramen ovale and no left atrial appendage clot. His Troponin-T was positive 1.2 ng/mL (normally less than 0.02 ng/mL). Haemoglobin was 19.5 g/dL and haematocrit of 61.4% s/o polycythemia. His platelet counts were initially 165×103 per cumm. There was no leukocytosis. Oxygen saturation was normal on Arterial Blood Gas (ABG) analysis. USG abdomen showed no splenomegaly. Noncontrast CT scan of the brain showed hyper density along the straight sinus, superior sagittal sinus, transverse sinus and sigmoid sinus suggestive of cerebral venous thrombosis [Table/Fig-1a,b]. MRI of the brain showed loss of flow void in the superior sagittal sinus, left transverse and sigmoid sinus s/o cerebral venous thrombosis [Table/Fig-1c]. Coronary angiogram was done in view of MI and it showed proximal PDA thrombotic 100% occlusion [Table/Fig-2a].
a,b) NCCT brain showed hyper density along straight sinus, superior sagittal sinus, transverse sinus and sigmoid sinus s/o cerebral venous thrombosis.
c) MRI brain showed loss of flow void in the superior sagittal sinus, left transverse and sigmoid sinus s/o cerebral venous thrombosis..
a) Coronary angiography showed proximal PDA thrombotic 100% occlusion; b) Thrombus aspiration was done following which TIMI 3 flow was obtained. Balloon dilation to proximal PDA branch was done.
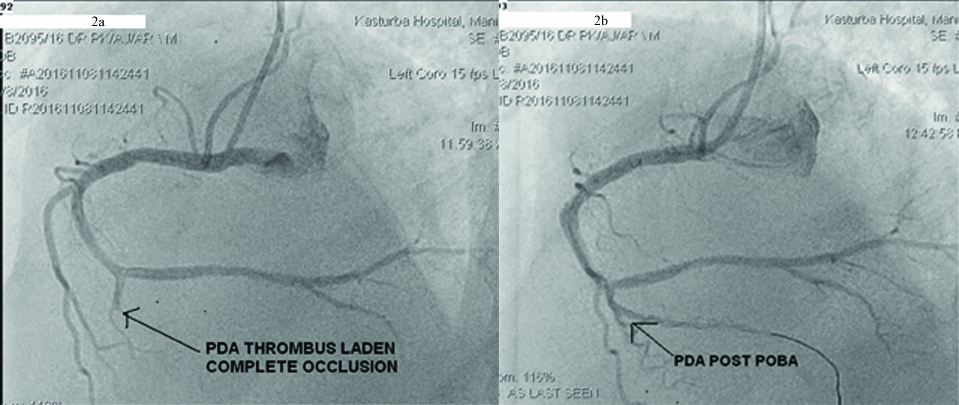
Thrombus aspiration was done following which Thrombolysis in MI (TIMI) 3 flow was obtained. Only balloon dilation to PDA was done alone as there was no stentable lesion [Table/Fig-2b]. Post procedure patient was started on single antiplatelet agent aspirin and oral anticoagulation with warfarin. C-ANCA, P-ANCA, and anti cardiolipin antibodies were negative. Phlebotomy was done in view of polycythemia and patient was discharged at a hematocrit of 50%. JAK2V617F gene was found to be negative and erythropoietin levels were normal (8 mU/mL, normal:4.1-19.5 mU/mL).
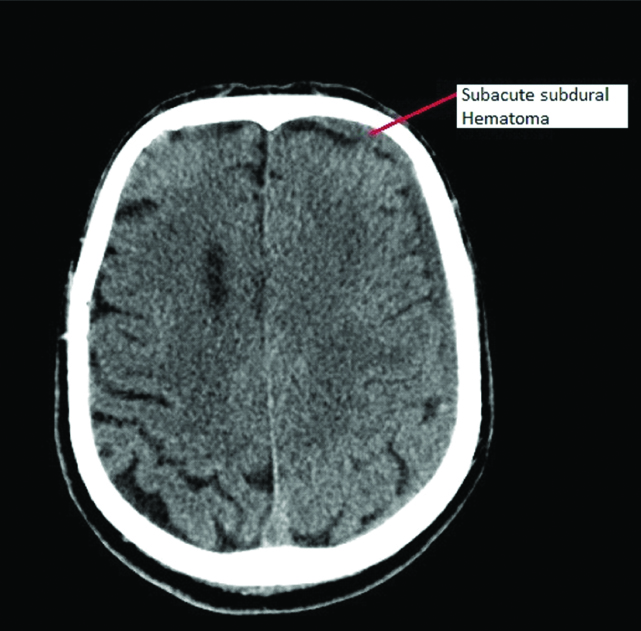
Nine days later, while on anticoagulation and Prothrombin Time (PT-INR) 1.56, patient presented to the hospital with sudden onset weakness of right upper and lower limb. CT scan of the brain showed subdural haematoma along left frontal temporal parietal convexity measuring 6.5 mm with a 6 mm midline shift to the right [Table/Fig-3]. The platelet counts were 87×103per cumm. Prothrombin time was five seconds. Fibrinogen levels were 1.2 g/dL. Fibrinogen degradation product levels were normal. His Disseminated Intravascular Coagulation (DIC) score was three. Bilateral carotid and vertebral arterial doppler were normal.
NCCT Brain was done which showed subdural haematoma along left frontal temporal parietal convexity measuring 6.5 mm with a 6 mm midline shift to the right.
Paroxysmal Nocturnal Haemoglobinuria (PNH) flow cytometry panel was done where immune phenotypic analysis was performed, using gating antibodies CD5 and GPI-linked protein Fluorescent Aerolysin (FLAER). Flow results showed no evidence of decreased or absent expression of FLAER in granulocytes and monocytes. It was concluded no phenotypic evidence of PNH. Haemoglobin was still high, 18.2 gm and haematocrit 55%.
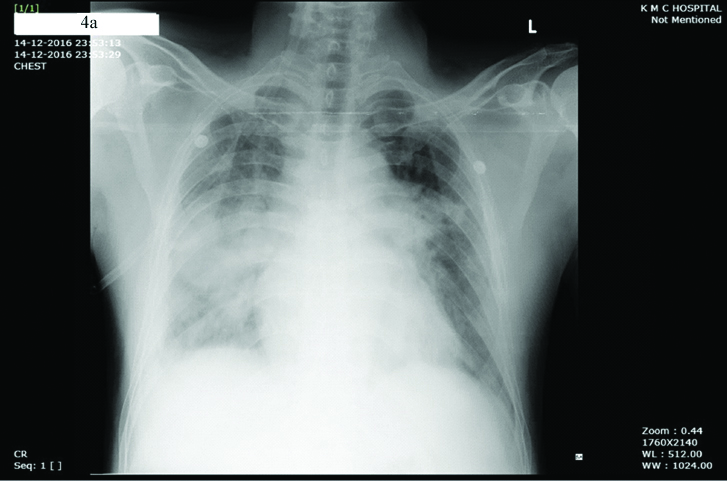
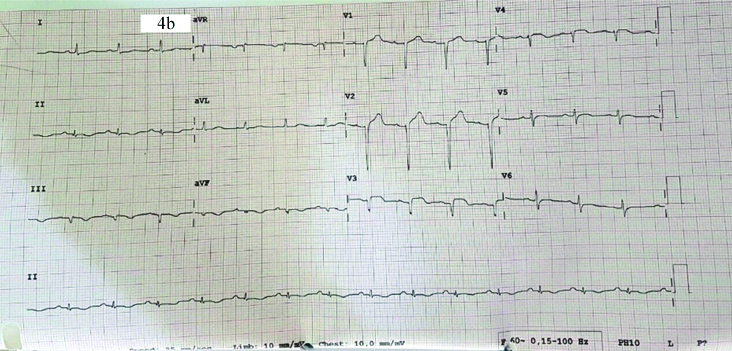
Antiplatelet and anticoagulation agents were stopped in view of acute bleed and patient’s weakness resolved. The patient improved and was discharged after a week with single antiplatelet. Two weeks later the patient had an episode of sudden severe chest pain associated with breathlessness and orthopnea at rest. He had bilateral basal crepitation in the chest and chest X-ray showed bilateral perihilar opacities suggesting pulmonary oedema and was hypoxic at rest [Table/Fig-4a]. His ECG showed evolved anterior wall MI with ST elevation and QS complexes in V1 to V4. [Table/Fig-4b]. Echocardiogram showed Left Anterior Descending (LAD) coronary artery territory hypokinesia with mild left ventricular systolic dysfunction. Coronary angiogram showed slow flow in LAD with again no stentable lesion [Table/Fig-4c]. Laboratory reports showed a haemoglobin of 15.2 g/dL and haematocrit of 46%. PT-INR was 1.04. Investigations revealed normal serum homocysteine (11.79 μmol/L, normal: 5.6-16.20 μmol/L), protein C (88.50%, normal: 70-140%), antithrombin III (93.10%, normal: 80-120%), erythropoietin (8 mU/mL, normal: 4.1-19.5 mU/mL) levels. Prothrombin gene mutation (G20210A), factor V Leiden mutation (R506Q), MTHFR (C677T) were all ruled out. However, as the protein S was found to be deficient (11.20%, normal:60-130%), it was considered as a hereditary deficiency of Protein S. In view of JAK negative PV and hereditary Protein S deficiency, patient was restarted on single antiplatelet and oral anticoagulation and hydroxyurea to prevent any further thrombotic episodes. Bone marrow examination was not done as patient was on antiplatelet agents as well as on oral anticoagulation.
Chest X-ray showed bilateral perihilar opacities suggesting pulmonary oedema.
ECG showed evolved anterior wall MI with ST elevation and QS complexes in V1 to V4.
Coronary angiography showing slow flow in LAD with no stentable lesion suggestive of reanalysed LAD.

Discussion
The PV is a rare disorder. It has a minimum incidence of 2.6 per 100000 [1]. The mutation of gene JAK2V617F is present in approximately 95% of patients with PV [2]. PV is a chronic myeloproliferative disorder with a high risk of both arterial and venous thromboembolic complication. Bleeding, thrombotic, and vascular complications occur in 40-60% of such patients resulting in increased morbidity and mortality. Hereditary protein S deficiency can not only cause venous thrombosis but is also a risk factor for arterial thrombosis including MI and stroke.
However, in some patients with classical PV, this gene mutation of JAK2V617F may be lacking. There is no significant difference in the clinical presentation of the PV in JAK2 positive and negative patients.
The diagnosis of PV is made according to the British Committee for Standards in Haematology (BCSH) guidelines. This patient satisfied the criteria for JAK negative PV as he had high haematocrit (>0.52 in men), with no mutation in JAK2, no secondary erythrocytosis, had no clinical as well as radiological evidence of splenomegaly.
Schafer AI, reported mortality due to thrombosis in 41% of 749 patients with a diagnosis of PV, thrombosis being directly responsible for death in 29% of cases [3]. Thrombotic complications, like MI, remain the primary cause of mortality and have been explained by increased whole blood viscosity, quantitative and qualitative platelet abnormalities and leukocytosis [3]. Despite the association of PV with coronary artery disease, the presentation of PV as AMI is very rare [4].
The pathophysiology of thromboembolic events has not been fully explained in PV, but many factors are implicated. There us increase in the haematocrit along with blood hyperviscosity, activation of platelet aggregation, thrombogenesis, leukocytosis and intimal proliferation. Increased haematocrit level is associated with increased blood viscosity, leading to decreased cerebral blood flow rate and increase in the hypercoagulable tendency in PV. In addition to increased blood viscosity, the axial migration of red cells leading to the displacement of platelets to the mural plasmatic zone occurs, which exposes them to maximal vessel wall shearing stress. Erythrocytosis increases the platelet-vascular interactions, particularly in arterioles and capillaries with high shear rates. This increased red cell mass in PV may also result in increased platelet activation.
Cortical venous thrombosis is also a rare but serious complication of polycythemia [5]. The present case also presented initially with a headache and found to have cerebral venous thrombosis on CT Brain.
The PV can cause both bleeding and thrombosis at different times in the same patient. Platelet abnormalities also contribute to bleeding at any site in such patients. In these patients, a subgroup of abnormally large platelets exists which is undergoing partial activation. This activation may be a result of collisions amongst these large platelets, forming microaggregates in the circulation. This phenomenon called disseminated intravascular platelet aggregation, may explain the in situ thrombosis in areas like coronary arteries with atherosclerosis with low flow. Also, recurrent aggregation and disaggregation of platelet microemboli in the circulation may impair the release function of the platelets [6].
This may explain why this patient developed subdural haematoma despite PT-INR being low suggesting that platelet function defects contribute to bleeding tendencies in PV.
A prior history of thrombosis, as seen in the present case, has been found to raise the risk of recurrent thrombosis in PV. In the ECLAP study, in patients with a history of thrombosis, the incidence of cardiovascular complications was higher than in those with no history of thrombosis [7]. The present case had a past history of deep vein thrombosis and subsequently developed cortical vein thrombosis and repeated episodes of MI.
Congenital Protein S deficiency is an autosomal dominant disease. Approximately 2% of patients with venous thromboembolism are found to be heterozygous for the same. Acquired Protein S deficiency is seen with warfarin therapy in the initial few days, liver disease, DIC, oral contraceptives, oestrogen therapy, acute phase inflammatory responses, pregnancy, HIV and sickle cell disease. None of these conditions were present in the present case, to have caused a Protein S deficiency state.
Protein S deficiency is a novel risk factor responsible for coronary atherosclerosis. Even under warfarin therapy, recurrent micro thrombi might form in the coronary arteries and lead to the progression of coronary atherosclerosis Bucalossi A et al., found a reduced concentration of natural anticoagulants (protein C), protein S and Antithrombin III in PV [8]. A total of the 57 % of patients showed one or more thrombotic episodes either at the diagnosis or during follow up. Natural anticoagulants were deficient in 43.5% of patients with a history of thrombosis versus only 5.7% in the group of patients without thrombosis.
However, in these patients, Protein S deficiency was found to be associated with Protein C deficiency and was not isolated, unlike this case where Protein C levels were found to be normal.
In the present case, no family history of thrombosis was present. This is common in a hereditary deficiency of Protein S. So it is open to discussion, whether the Protein S deficiency in the present case, was genetic or whether it was due to PV.
Also, the reduction of haematocrit after phlebotomy did not eliminate the thrombotic risk. The patient had another episode of MI, probably due to the additional Protein S deficiency.
Long term management with low dose aspirin in PV patients has been shown to reduce the rate of major thrombosis as well as cardiovascular death and is recommended in all patients in the absence of contraindications [7].
Willoughby S and Pearson TC, reviewed 86 studies and concluded that prophylactic aspirin use will reduce the occurrence of major vascular events by 22% and nonfatal MI by 30% in patients with PV [9].
De Stefano V et al., have also reported antiplatelet agents and anticoagulants can independently prevent recurrences, reduce rethrombosis by 58% and 68% respectively [10]. However, no data exists for dual antiplatelet therapy of aspirin with a thienopyridine. Also, no established guidelines are there for treating patients with ACS or STEMI and PV. Phlebotomy and close haemodynamic monitoring are suggested as an additional approach to standard treatments.
In a patient with PV, an acute thrombosis is managed with heparin and oral anticoagulant therapy. Systemic anticoagulation alone may not be enough and concomitant myelosuppression with hydroxyurea is highly recommended. Cytoreduction of blood hyper viscosity by phlebotomy or chemotherapy with low dose aspirin have dramatically decreased thrombotic complications and markedly improved survival.
Although, there are no current guidelines for treatment of acute MI in PV patients, current evidence supports the standard antiplatelet therapy along with the use of hydroxyurea as the initial approach in PV patients with the acute coronary syndrome [11].
Attending physicians should have a high index of suspicion for PV and Protein S deficiency, especially when a young person presents with MI in the absence of usual atherosclerotic risk factors, as the initiation of early management will alter patient prognosis.
Conclusion
This rare case illustrates the importance of diagnosing PV and Protein S deficiency as an important cause of thrombosis including MI and to emphasise that early recognition of the disease will help to consider appropriate management strategies.
[1]. Tefferi A, Spivak JL, Polycythemia vera: scientific advances and current practiceSemin Hematol 2005 42(4):206-20.10.1053/j.seminhematol.2005.08.00316210034 [Google Scholar] [CrossRef] [PubMed]
[2]. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disordersLancet 2005 365(9464):1054-61.10.1016/S0140-6736(05)71142-9 [Google Scholar] [CrossRef]
[3]. Schafer AI, Molecular basis of the diagnosis and treatment of polycythemia vera and essential thrombocythemiaBlood 2006 107(11):4214-22.10.1182/blood-2005-08-352616484586 [Google Scholar] [CrossRef] [PubMed]
[4]. Bahbahani H, Aljenaee K, Bella A, Polycythemia vera presenting as acute myocardial infarction: An unusual presentationJ Saudi Heart Assoc 2015 27(1):57-60.10.1016/j.jsha.2014.07.00325544823 [Google Scholar] [CrossRef] [PubMed]
[5]. McBane RD, Tafur CAC, Wysokinski WE, Acquired and congenital risk factors associated with cerebral venous sinus thrombosisThromb Res 2010 126(2):81-87.10.1016/j.thromres.2010.04.01520541240 [Google Scholar] [CrossRef] [PubMed]
[6]. Mehta P, Mehta J, Ross M, Ostrowski N, Player D, Decreased platelet aggregation but increased thromboxane A2 generation in polycythemia veraArchives of internal medicine 1985 145(7):1225-27.10.1001/archinte.1985.003600701030164015270 [Google Scholar] [CrossRef] [PubMed]
[7]. Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, Efficacy and safety of low-dose aspirin in polycythemia veraN Engl J Med 2004 350(2):114-24.10.1056/NEJMoa03557214711910 [Google Scholar] [CrossRef] [PubMed]
[8]. Bucalossi A, Marotta G, Bigazzi C, Galieni P, Dispensa E, Reduction of antithrombin III, protein C, and protein S levels and activated protein C resistance in polycythemia vera and essential thrombocythemia patients with thrombosisAm J hematol 1996 52(1):14-20.10.1002/(SICI)1096-8652(199605)52:1<14::AID-AJH3>3.0.CO;2-9 [Google Scholar] [CrossRef]
[9]. Willoughby S, Pearson TC, The use of aspirin in polycythemia vera and primary thrombocythaemiaBlood Reviews 1998 12(1):12-22.10.1016/S0268-960X(98)90026-1 [Google Scholar] [CrossRef]
[10]. De Stefano V, Za T, Rossi E, Vannucchi AM, Elli M, Sheridan E, Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatmentsHaematologica 2008 93(3):372-80.10.3324/haematol.1205318268279 [Google Scholar] [CrossRef] [PubMed]
[11]. Adel G, Aoulia D, Amina Y, Aymen BA, Abdel-Hamid NM, Polycythemia Vera and acute coronary syndromes: pathogenesis, risk factors, and treatmentJ Hematol Thromb Dis 2013 15(2):02-05. [Google Scholar]