Aortopulmonary Window: Case Report of Survival in Untreated Adult Patient
Neha Nischal1, Sakshi Arya2, Ruchi Gupta3, Nayna Goyal4, Sunil Kumar Puri5
1 Senior Resident, Department of Radiology, GB Pant Hospital, New Delhi, Delhi, India.
2 Senior Resident, Department of Radiology, GB Pant Hospital, New Delhi, Delhi, India.
3 Senior Resident, Department of Radiology, AIIMS, Patna, Bihar, India.
4 Senior Resident, Department of Radiology, GB Pant Hospital, New Delhi, Delhi, India.
5 Director Professor and Head, Department of Radiology, GB Pant Hospital, New Delhi, Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Neha Nischal, 1, JLN Marg, New Delhi-110002, Delhi, India.
E-mail: neha.nischal@gmail.com
Aortopulmonary window is a rare congenital communication between ascending aorta and main pulmonary artery. The condition carries poor prognosis in absence of early corrective surgery. Few case reports exist in literature where adult survival is seen in untreated patients. We present such a case of a 27-year-old male who had developed irreversible pulmonary hypertension secondary to aortopulmonary window and is thus being managed medically.
Aortopulmonary septal defect, Echocardiography, Eisenmenger, Pulmonary hypertension
Case Report
A 27-year-old male patient with a history of dyspnoea on exertion (NYHA grade II/III) and two episodes of haemoptysis was admitted in the cardiology department of our hospital. The patient suffered from recurrent respiratory infections since childhood which required repeated hospital admissions and was subsequently diagnosed with an underlying cardiac pathology for which surgery had been advised. However, the parents were not willing for the surgery. The exact diagnosis was not made as previous records were not available with the patient. Over a period of time, the patient’s symptoms had subsided with normal growth.
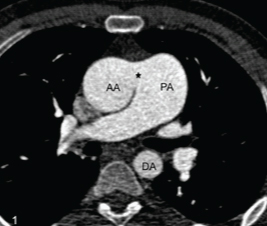
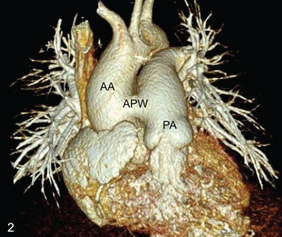
On physical examination, the patient showed pan digital clubbing. On auscultation, there was a aloud S2 with a short ejection systolic murmur along the left lower sternal border in the second intercostal space. The oxygen saturation of the patient was 80-85% at rest, which showed further desaturation with exercise. Echocardiography revealed biventricular hypertrophy. On transthoracic echocardiography, there was a mild dilatation of right atrium, right ventricle and pulmonary arteries with mild tricuspid regurgitation. The right ventricular systolic pressure was 110 mmHg by Tricuspid Regurgitation (TR) jet with reduced ejection fraction (30-35%). Contrast echocardiography revealed opacification of ascending aorta with agitated saline. However, no atrial or Ventricular Septal Defect (VSD) could be demonstrated thus confirming the presence of a right to left shunt. Chest X-ray revealed mild bilateral hilar prominence with no other remarkable finding. The patient was referred to the radiology department for CT angiography for evaluation of the size of the defect as well as for other cardiac and extracardiac anomalies. CT showed a large abnormal communication (measuring approximately 17 mm) between the left wall of ascending aorta and the right wall of main pulmonary artery [Table/Fig-1,2]. The main pulmonary artery as well as the right and left pulmonary arteries were mildly dilated with peripheral pruning of pulmonary vasculature. No other associated cardiac anomaly was seen. Aortic arch was left sided with normal branching pattern, normal origin and course of the coronary arteries. Lung parenchyma was normal. A final diagnosis of aortopulmonary window with Eisenmenger syndrome was made. In view of irreversible pulmonary hypertension, surgery was contraindicated and thus the patient is being managed with sildenafil.
Axial CECT at the level of ascending aorta shows a large communication (*) between the ascending aorta (AA) and the pulmonary artery (PA) with dilated PA.
A 3D VR image depicts the type i aorto-pulmonary window (APW) between the ascending aorta (AA) and pulmonary artery (PA) with two separate semilunar valves.
Discussion
Aortopulmonary septal defect is an uncommon congenital anomaly accounting for less than 1% of all congenital heart diseases [1]. It is an abnormal communication between the ascending aorta and main pulmonary artery resulting from a deficient septum between the two. Currently, it is classified into four types: Type I (proximal)- located just above the sinus of Valsalva; Type II (distal)-defect in upper portion of the ascending aorta; Type III (total)-involvement of the entire aortopulmonary septum; Type IV (intermediate)-these defects have adequate superior and inferior rims and they are most suitable for possible device closure [2].
This defect may be isolated or may be associated with other congenital cardiac anomalies including patent ductus arteriosus (most common), interrupted aortic arch, Atrial Septal Defect (ASD), VSD, coronary artery anomalies in about half of the patients [3].
It is considered in the spectrum of conotruncal anomalies with truncus arteriosus at one end and aortopulmonary window at the other. An important difference between the two is that the aortic and pulmonary valves are separate in aortopulmonary window whereas both aorta and pulmonary artery arise together from a common semilunar valve in truncus arteriosus [4].
The condition manifests soon after birth with gradual decline in the pulmonary vascular resistance over the first few days of life resulting in the development of a left to right shunt. This leads to pulmonary oedema and symptoms of congestive heart failure similar to those seen with other congenital left to right cardiac shunts. If left untreated, progression to pulmonary vascular obstructive disease is bound to occur because of increasing pulmonary pressures, with ultimate reversal of the shunt [5].
Symptoms of congestive heart failure and early pulmonary hypertension along with relevant clinical investigations should raise a strong suspicion for this anomaly. Echocardiography and cross-sectional imaging are important in diagnosing this condition by depicting the defect clearly. The importance of colour Doppler study cannot be undermined as it quantifies the flow mechanics across the defect and is thus helpful in assessing its size. Cardiac catheterisation remains the gold standard for diagnosing the defect with evaluation of the shunt [3,5]. However, cross-sectional imaging including CT and MRI may suffice in many patients by depicting the defect clearly as well as evaluation of lung parenchyma and other related anomalies. This can obviate the need for an invasive procedure, as in present case, where the patient was not a surgical candidate [6].
Surgical closure of the defect is indicated as soon as the diagnosis is made and carries a good prognosis in absence of other anomalies. Left untreated, it carries a high mortality of about 40% in infancy, this being the reason of only a few cases of untreated adult survival being reported [1,7]. The median age of survival in such patients is 33 years [3]. The longest reported survival is of a female who, although symptomatic, had a preserved quality of life and died at the age of 60 years [8]. Even in adults, if the patient presents before development of Eisenmenger syndrome, corrective surgery can be done. If irreversible pulmonary hypertension has developed, patient is usually managed with anticoagulants and phlebotomy if haematocrit is more than 55% [3].
Conclusion
Aortopulmonary window is a very rare congenital cardiac anomaly and adult survival without treatment, although known, is quite uncommon. Our patient, 27 years of age, is being managed medically in view of having irreversible pulmonary hypertension. Imaging with CT along with relevant clinical findings sufficed in our case and cardiac catheterisation was not required. This anomaly should be kept in mind and sought for in an adult patient with unexplained pulmonary hypertension.
[1]. Yüksel IO, Kğklü E, Arslan S, Üreyen CM, Küçükseymen S, Aortopulmonary window in adulthood: Surviving at 22 years without intervention or pulmonary vascular diseaseTurk Kardiyol Dern Ars 2016 44(4):332-34.10.5543/tkda.2016.9422427372619 [Google Scholar] [CrossRef] [PubMed]
[2]. Backer CL, Mavroudis C, Surgical management of aortopulmonary window: a 40-year experienceEur J Cardiothorac Surg 2002 21(5):773-79.10.1016/S1010-7940(02)00056-8 [Google Scholar] [CrossRef]
[3]. Kose M, Ucar S, Emet S, Akpinar TS, Yalin K, A case of aortopulmonary window: asymptomatic until the first pregnancyCase Rep Cardiol 2015 2015:93525310.1155/2015/93525326457208 [Google Scholar] [CrossRef] [PubMed]
[4]. Ghaderian M, Aortopulmonary window in infantsHeart Views 2012 13(3):103-06.10.4103/1995-705X.10215323181179 [Google Scholar] [CrossRef] [PubMed]
[5]. Demir IH, Erdem A, Sarıtas T, Demir F, Erol N, Yücel IK, Diagnosis, treatment and outcomes of patients with aortopulmonary windowBalkan Med J 2013 30(12):191-96.10.5152/balkanmedj.2013.699525207099 [Google Scholar] [CrossRef] [PubMed]
[6]. Sridhar PG, Kalyanpur A, Suresh PV, Sharma R, Maheshwari S, Hrudayalaya N. Helical CT evaluation of aortopulmonary septal defectInd J RadiolImag. 2006 16(4):1847-49.10.4103/0971-3026.32364 [Google Scholar] [CrossRef]
[7]. Nadig S, Kapoor A, Kumar S, Gaharwar S, Phadke RV, A rare case of large aortopulmonary window with Eisenmenger syndrome and adult survivalJournal of Cardiology Cases 2014 10(5):193-95.10.1016/j.jccase.2014.07.00830534241 [Google Scholar] [CrossRef] [PubMed]
[8]. Su-Mei AK, Ju-Le T, Large un-repaired aortopulmonary window-survival into the seventh decadeEchocardiography 2007 24(1):71-73.10.1111/j.1540-8175.2006.00353.x17214626 [Google Scholar] [CrossRef] [PubMed]