Systemic Lupus Erythematosus Presenting as Macrophage Activation Syndrome
Satyabrata Guru1, Anupama Behera2, Nishant Debta3, Rajesh Kumar4
1 Assistant Professor, Department of Trauma and Emergency Medicine, AIIMS, Bhubaneswar, Odisha, India.
2 Assistant Professor, Department of Medicine, AIIMS, Bhubaneswar, Odisha, India.
3 Ex-Senior Resident, Department of Medicine, AIIMS, Bhubaneswar, Odisha, India.
4 Assistant Professor, Department of Medicine, AIIMS, Bhubaneswar, Odisha, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Satyabrata Guru, QRT-V(S)-2, Infront of Pathani Samant Hostel, Ouat Colony, Bhubaneswar-751003, Odisha, India.
E-mail: satyabrataguru@yahoo.com
Macrophage Activation Syndrome (MAS), a potentially life threatening condition belongs to acquired cause of Hemophagocytic lymphohistiocytosis (HLH) group of diseases more often found in children than adults. It presents as familial forms or acquired forms that include infectious aetiologies notably viral, malignancies, drugs and autoimmune diseases. Its association with several systemic autoimmune disease includes commonly systemic Juvenile Idiopathic Arthritis (sJIA), adult-onset Still’s disease and rarely with Systemic Lupus Erythematosus (SLE), rheumatoid arthritis and dermatomyositis. It is a heterogeneous presentation of systemic and laboratory findings characterised by abnormal immune activation and inflammation warrants early diagnosis and prompt treatment. Herein we report a rare case of 15-year-old female who presented with fever of two months duration, who was later diagnosed to be having MAS with SLE. Despite the rarity MAS can be initial presenter in SLE and should be searched in the background of fever and features of SLE. Initiation of treatment with pulse methylprednisolone and follow up with oral hydroxychloroquine, cyclosporine and steroids had a favourable outcome.
Polymerase chain reaction, Raynaud’s phenomenon, Hyperferritinaemia
Case Report
A 15-year-old female was referred to our hospital (AIIMS) with complaint of intermittent fever since two months with no prior history of arthritis, photosensitivity, mouth ulceration, rash or Raynaud’s phenomenon. She had received treatment with multiple broad spectrum antibiotics, antifungals but fever did not subside.
Our clinical examination showed malar rash, non painful oral ulceration, arthritis involving small and large joints of upper and lower limbs with stable vitals.
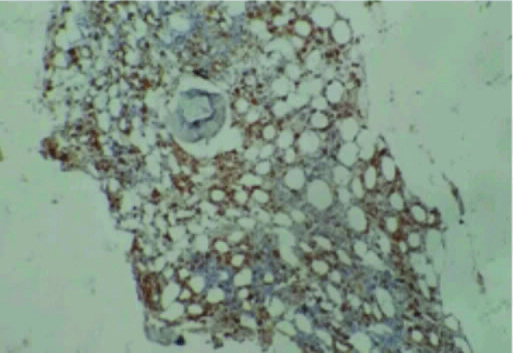
Investigations revealed pancytopenia Hb- 9.1 gm/dL (range:12-15.8 gm/dL), TLC-2100/mm3 (range:3540-9060/mm3), platelet-45,000/mm3 (range:165000-415000/mm3)}, ESR-35 mm/hour (range: 0-20 mm/hour), 24 hour urine protein- 240 mg/dL (range: <150 mg/dL). Absence of malarial parasite and antigen, negative typhidot, negative serology for scrub typhus, HIV, hepatitis B and C. Viral polymerase chain reaction for cytomegalovirus, epstein barr virus, herpes simplex virus, parvovirus B19 was negative, repeated absence of bacterial and fungal growth in blood, urine and bone marrow, negative tuberculin senstivity test. Liver function test revealed elevated transaminase levels (serum Bilirubin-1.4 mg/dL (range: 0.3-1.3 mg/dL), AST- 202 U/L (range: 12-38 U/L), ALT- 205 U/L (range: 7-41 U/L), ALP- 667 IU/L (range: 44-147 U/L), Immunological tests included – ANA-Positive (1:240-homogeneous pattern), Anti dsDNA (>800 UI/mL) and anti-Sm (158 UI/mL), low complements (C3- 0.1 gm/L and, C4- 0.9 gm/L), Positive (3+) direct coombs test, low fibrinogen- (28 mg/dL), high serum ferritin (>15000 ng/mL), increased triglycerides (592 mg/dL). CT-abdomen revealed mild hepatosplenomegaly, mild ascitis and CT of thorax showed mild bilateral pleural effusion. Bone marrow biopsy showed hypocellularity, dyspoiesis, and increased histiocytes with hemophagocytosis [Table/Fig-1,2].
Trephine biopsy showing hypocellularity and increased number of histiocytic cells and engulfed cells (CD68: IHC, 20X).
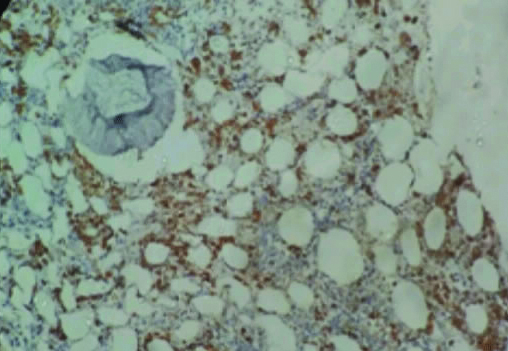
Trephine biopsy showing increased CD68+ cells (histiocytic) with hemophagocytosis (IHC, 40X).
Above findings were suggestive of SLE presenting as MAS. Patient was given pulse intravenous methylprednisolone (1 gm/day for three days), followed by oral steroids, hydroxychloroquine and oral cyclosporine and regular follow up after one month showed complete subsidence of symptoms of the disease and reversal of abnormal parameters.
Discussion
Macrophage activation syndrome is a severe complication of rheumatic disease that has rapid fatal outcome involving multiple organs thus requiring early recognition and treatment. Familial HLH is more common in children due to genetic association. Triggering factors in secondary HLH includes infections, malignancy, drugs and rheumatic disorder [1]. Among rheumatic disorders, sJIA is most common cause followed by its adult counterpart. The incidence of MAS associated with SLE is rare accounting to 0.9-4.6% [2]. It is characterised by fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, hyperferritinaemia, hypertriglyceridemia, hypofibrinogenemia and hemophagocytosis. In any SLE patient presenting with clinical signs of disease flare, MAS should be included in the differential diagnosis. High serum ferritin and normal ESR levels may differentiate between MAS and SLE flare but all features must be considered and rapid evaluation is required [3]. The pathogenesis includes cytotoxic cell killing and cytokine storm. MAS is characterised by intense systemic inflammatory reaction caused by a massive dysregulation of macrophage- lymphocyte interactions (increase TNF-α, M-CSF receptors, IL-1, IL-6, and IFN-α) [4]. Abnormal clearance and activation of macrophages leads to hemophagocytosis (engulfement of host blood cells) causing pancytopenia as was evident here in bone marrow biopsy. The presence of polyarthritis, malar rash, polyserositis, positive ANA and anti dsDNA suffices to reach at a diagnosis of SLE by Systemic Lupus Collaborating Clinics (SLICC) criteria [5]. Ruling out infectitious aetiology was a pointer towards alternative diagnosis. The findings of hyperferritenemia, hypofibrinogenemia, hypertriglyceridemia, pancytopenia, raised transaminases and hemophagocytosis were suggestive of MAS as per HLH-2004 diagnostic and therapeutic guide lines [1]. Establishing the diagnosis of MAS is a challenging procedure because the clinical features of MAS-associated SLE and active SLE are very similar. However, hyperferritinemia is the best parameter to discriminate between MAS-associated SLE and active SLE with a sensitivity and specificity of almost 100% [6]. MAS associated SLE is a known fact but MAS as presenter of SLE is rare. Till date, only 26 cases of MAS only related to the onset of SLE are reported in the literature [7]. Because of its potential fatal nature high index of suspicion is necessary. Treatment includes high-dose glucocorticosteroids and second-line therapy based on cyclosporine, intravenous immunoglobulins, etoposide and recently biologicals in non responders.
Conclusion
Although, MAS as initial presenter of SLE is rare and its diagnosis remains a challenge to physician because of its potential fatal nature and high index of suspicion is necessary. Any female patient with features of SLE with pyrexia of unknown origin and pancytopenia, MAS as initial presenter of SLE is an important differential in background of absence of infectitious aetiology.
[1]. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosisPediatric Blood and Cancer 2007 48(2):124-31.10.1002/pbc.2103916937360 [Google Scholar] [CrossRef] [PubMed]
[2]. Vilaiyuk S, Sirachainan N, Wanitkun S, Pirojsakul K, Vaewpanich J, Recurrent macrophage activation syndrome asthe primary manifestation in systemic lupus erythematosus and the benefit of serial ferritin measurements: a case-based reviewClinical Rheumatology 2013 32(6):899-904.10.1007/s10067-013-2227-123483294 [Google Scholar] [CrossRef] [PubMed]
[3]. Levy DM, Kamphuis S, Systemic lupus erythematosus in children and adolescentsPediatric Clinics of North America 2012 59(2):345-64.10.1016/j.pcl.2012.03.00722560574 [Google Scholar] [CrossRef] [PubMed]
[4]. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, Cytokine production regulating Th1 andTh2 cytokines in hemophagocyticlymphohistiocytosisBlood 1997 89(11):4100-03.10.1182/blood.V89.11.41009166851 [Google Scholar] [CrossRef] [PubMed]
[5]. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin FR, Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosusArthritis Rheum 2012 64(8):2677-86.10.1002/art.3447322553077 [Google Scholar] [CrossRef] [PubMed]
[6]. Granata G, Didona D, Stifano G, Feola A, Granata M, Macrophage Activation syndrome as onset of systemic lupus erythematosus: a case report and a review of the literatureCase Reports in Medicine 2015 (2015):01-04.10.1155/2015/29404126064125 [Google Scholar] [CrossRef] [PubMed]
[7]. Carvalheiras G, Anjo D, Mendonça T, Vasconcelos C, Farinha F, Hemophagocytic syndrome as one of the main primary manifestations in acute systemic lupus erythematosus-case report and literature reviewLupus 2010 19(6):756-61.10.1177/096120330935490620026522 [Google Scholar] [CrossRef] [PubMed]