Myoclonic Seizures and Lactic Acidemia in a Term Newborn Born with No History of Asphyxia
Sushil Choudhary1, Viraraghavan Vadakkencherry Ramaswamy2, Sushma Nangia3, Arvind Saili4
1 Resident, Department of Neonatology, Lady Hardinge Medical College, New Delhi, India.
2 Resident, Department of Neonatology, Lady Hardinge Medical College, New Delhi, India.
3 Director Professor, Department of Neonatology, Lady Hardinge Medical College, New Delhi, India.
4 Director Professor, Department of Neonatology, Lady Hardinge Medical College, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Viraraghavan Vadakkencherry Ramaswamy, 4th Floor, 1176, Sat Nagar, Karol Bagh-110005, New Delhi, India.
E-mail: 19.vira@gmail.com
We here report an unusual case of a term neonate who presented with myoclonic seizures and lactic acidemia at 30 hours of life, who was later on diagnosed as a case of pyridoxine dependant epilepsy. Pyridoxine dependant epilepsy is one of the rare but easily treatable causes of refractory neonatal seizures. Failure to diagnose and treat this condition early would result in devastating neurodevelopmental outcomes for the surviving newborn.
Burst suppression pattern, Epilepsy, Neonatal metabolic acidemia, Refractory neonatal seizures
Case Report
A term neonate had presented to the Neonatal Intensive Care Unit (NICU) at 30 hours of life with altered sensorium and multiple episodes of myoclonic seizures. He was a product of third degree consanguineous marriage. There was a significant history of early neonatal death in the elder sibling on day 4 of life which was not properly investigated. The baby was born vaginally at 38 weeks and had cried immediately after birth with normal APGAR scores of 8 and 9 at one and five minutes respectively. He was shifted with the mother to the maternity ward at eight hours of life and was exclusively breast fed since birth.
At 30 hours of life the newborn presented to the NICU with multiple episodes of myoclonic seizures. The blood dextrose was normal. A dose of calcium gluconate was given empirically at 2 mL per kg after drawing a blood sample. The seizures failed to abort and he was loaded with injection phenobarbitone followed by injection phenytoin subsequently. In view of consanguinity and a positive family history, a strong suspicion of an inborn error of metabolism was kept. The baby was started on intravenous fluids and kept nil per oral. The seizures subsequently aborted at around 36 hours of life. Blood investigations for sepsis and various other metabolic derangements were performed. The initial evaluation revealed lactic acidemia with a pH of 7.15 (Normal level 7.35-7.45) and lactate levels of 20 mmol/L (Normal level 0.5-1.5 mmol/L). Tandem Mass Spectrometry (TMS) and urine Gas Chromatography Mass Spectroscopy (GCMS) reports were awaited.
MRI and Magnetic Resonance Spectroscopy (MRS) brain was done to rule out any malformations and various other Inborn errors of metabolisms with lactic acidemias and was normal. EEG was done on the 3rd day of life and revealed a burst suppression pattern [Table/Fig-1]. The provisional diagnosis was narrowed down to glycine encephalopathy, pyridoxine dependant seizures or any of the progressive neonatal encephalopathy syndromes. The anticonvulsants were slowly tapered off and a cocktail therapy with pyridoxine, folinic acid, biotin, Vitamin B12 and thiamine was started. The blood pH and lactic academia progressively improved and normalised by 4th day of life.
EEG revealing a burst suppression pattern on day 2.
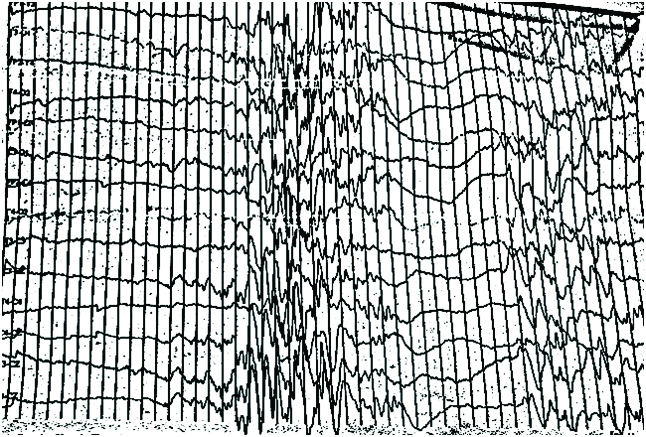
Subsequently the TMS and GCMS reports came out to be negative. All the other drugs except pyridoxine was withdrawn. The baby was started on minimal enteral nutrition on the 5th day of life which was subsequently hiked. He tolerated the feeds well and there was a dramatic improvement in his sensorium over a period of next two days. A repeat EEG was done 72 hours after the first EEG on 7th day of life which showed a normal pattern, confirming the electrographic response to oral pyridoxine supplementation [Table/Fig-2].
Normalisation of the burst suppression pattern on day 5.

At the time of the discharge, the baby was feeding directly from the breast with a normal neurological examination. The parents were referred to genetic counselling. The newborn was followed up at three months of post natal age. He had gained adequate weight and was being exclusively breast fed. Developmental assessment revealed isolated gross motor development delay with head control not being achieved. There was no recurrence of seizures. Plasma pipecolic acid levels were done on follow up while on pyridoxine supplementation by TMS and was found to be elevated 4 folds with a value of 6.21 umol/L {Normal value-Mean-1.46 μmol/L (range 0.70-2.46 μmol/L)}.
Discussion
Pyridoxine dependent epilepsy is a rare autosomal recessive disorder causing refractory seizures in neonatal period with an incidence of 1:400000 to 1: 7500000 [1]. Mutation in ALDH7A1 gene is the most common cause for pyridoxine dependent epilepsy [2]. This genetic defect causes a deficiency in enzyme Alpha Aminoadipic Semialdehyde (AASA) dehydrogenase, resulting in accumulation of AASA, Piperidine-6-Carboxylic Acid (P6C) and pipecolic acid [3]. The metabolite P6C inactivates the active form of pyridoxine (pyridoxyl 5 phosphate), which in turn is required for the synthesis of the inhibitory neurotransmitter GABA. Pyridoxine supplementation will compensate for the lost pyridoxal-5-phosphate.
Pyridoxine dependant epilepsy typically presents in the neonatal period as seen in our case [4]. This disorder should be suspected whenever a neonate born out of a consanguineous marriage presents with refectory seizures or when there is a previous sibling death with similar symptomatology. This typical history was present in our case. The most common type of seizures is generalised tonic or clonic seizures. Myoclonic seizures as seen in our case, though rare has also been reported previously [5,6]. The symptomatology can be varied ranging from developmental delay without seizures to behavioural disorders like autism [7]. Some children might present later after the infancy period also [8]. Lactic acidosis which was present in our case is not typically seen in pyridoxine dependant epilepsy. Mercimek-Mahmutoglu S, et al., had reported a case of a neonate who presented with hypoglycemia and lactic acidosis on 3rd day of life [9]. This baby unlike our case had hypoglycemia asso-ciated with lactic acidosis and was worked up for a disorder of gluconeogenesis. Response to pyridoxine further lead to the diagnosis of pyridoxine dependant epilepsy in this neonate.
MRI done in our case revealed a normal study. Different MRI findings have been reported such as thinning of the corpus callosum, mega cisterna magna, cerebellar atrophy, and various degrees of gray and white matter atrophy [10,11].
EEG done in our case revealed a burst suppression pattern. Nabbout R, et al., had described the various EEG changes seen in pyridoxine dependant epilepsy in a case series of nine patients [12]. Apart from the burst suppression pattern that was seen in three cases, Nabbout R, et al., had described a combination of continuous and discontinuous patterns in two cases and bilateral high voltage delta slow wave activity in four cases. The diagnosis can be quickly achieved by demonstrating normalisation of the various EEG abnormalities within minutes to days of treatment with pyridoxine [12]. It should be borne in mind that pyridoxine can cause apnoea and hypotension. Hence all this test should be done in a controlled environment. Even oral treatment with pyridoxine at 30 mg/kg/day for two to three days can be used to see for EEG improvement. Blood, urine and CSF levels of AASA, P6C and pipecolic acid can be elevated in this disorder [3]. We had done plasma pipecolic acid levels on follow up and was found to be four folds elevated despite treatment. Elevated levels of pipecolic acid are reported despite supplementation with pyridoxine [4].
Treatment is oral pyridoxine at a dose of 15 to 30 mg/kg/day. The treatment should be continued life long.
Conclusion
Pyridoxine dependant seizures should be kept as a differential diagnosis in any neonate presenting with refractory seizures with an absent history of birth asphyxia and with a positive family history. Any sudden death in a newborn baby should be properly investigated for inborn errors of metabolism to avert future disasters. The missed diagnosis in the previous sibling was disastrous to this baby. Pyridoxine dependant seizure presenting with lactic academia is a rare finding that was present in this case and this easily treatable disorder should be kept as a possibility before drawing a conclusion towards other rare mitochondrial disorders such as Leigh syndrome like diseases that might have a neonatal presentation.
[1]. Gospe SM, Pyridoxine-dependent seizures: new genetic and biochemical clues to help with di-agnosis and treatmentCurr Opin Neurol 2006 19(2):148-153.10.1097/01.wco.0000218230.81301.1216538088 [Google Scholar] [CrossRef] [PubMed]
[2]. Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Mutations in antiquitin in individuals with pyridoxine-dependent seizuresNat Med 2006 12(3):307-309.10.1038/nm136616491085 [Google Scholar] [CrossRef] [PubMed]
[3]. Plecko B, Paul K, Paschke E, Stoeckler-Ipsiroglu S, Struys E, Jakobs C, Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) geneHum Mutat 2007 28(1):19-26.10.1002/humu.2043317068770 [Google Scholar] [CrossRef] [PubMed]
[4]. Stockler S, Plecko B, Gospe SM, Coulter-Mackie M, Connolly M, van Karnebeek C, Pyri-doxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-upMol Genet Metab 2011 104(1-2):48-60.10.1016/j.ymgme.2011.05.01421704546 [Google Scholar] [CrossRef] [PubMed]
[5]. Mikati MA, Trevathen E, Krishnamoorthy KS, Lombroso CT, Pyridoxine-dependent epilepsy: EEG investigations and long-term follow-upElectroencephalography Clin Neurophysiol 1991 78(3):215-22.10.1016/0013-4694(91)90035-3 [Google Scholar] [CrossRef]
[6]. Krishnamoorthy KS, Pyridoxine-dependency seizures: Report of a rare presentationAnn Neu-rol 1983 13(1):103-04.10.1002/ana.4101301236830153 [Google Scholar] [CrossRef] [PubMed]
[7]. Burd L, Stenehjem A, Fraceschini LA, Kerbeshian J, A 15 year follow-up of a boy with pyri-doxine-dependent seizures with autism, breath holding and severe mental retardationJ Child NeuroI 2000 15(11):763-65.10.1177/08830738000150111111108513 [Google Scholar] [CrossRef] [PubMed]
[8]. Bachman DS, Late onset pyridoxine dependent convulsionAnn Neurol 1983 14(6):692-93.10.1002/ana.4101406186651254 [Google Scholar] [CrossRef] [PubMed]
[9]. Mercimek-Mahmutoglu S, Horvath GA, Coulter-Mackie M, Nelson T, Waters PJ, Sargent M, Profound neonatal hypoglycemia and lactic acidosis caused by pyridoxine-dependent epilepsyPediatrics 2012 129(5):1368-72.10.1542/peds.2011-012322529283 [Google Scholar] [CrossRef] [PubMed]
[10]. Baxter P, Griffiths P, Kelly T, Gardner-Medwin D, Pyridoxine dependent seizures: demographic, clinical, MRI and psychometric features, and effect of dose on intelligence quotientDev Med Child Neurol 1996 38(11):998-1006.10.1111/j.1469-8749.1996.tb15060.x8913181 [Google Scholar] [CrossRef] [PubMed]
[11]. Gospe SM, Hecht ST, Longitudinal MRI findings in pyridoxine-dependent seizuresNeurology 1998 51(1):74-78.10.1212/WNL.51.1.749674782 [Google Scholar] [CrossRef] [PubMed]
[12]. Nabbout R, SouZet C, Plouin P, Dulac O, Pyridoxine dependent epilepsy: a suggestive electroc-linical patternArch Dis Child Fetal Neonatal Ed 1999 8(2):F125-29.10.1136/fn.81.2.F12510448181 [Google Scholar] [CrossRef] [PubMed]