Idiopathic Non-Specific Interstitial Pneumonia (NSIP) is a rare entity in children and classified into cellular NSIP (c-NSIP) or fibrotic NSIP patterns. It comes under major idiopathic interstitial pneumonia. Both conditions are characterised by Ground Glass Opacities (GGO), irregular linear opacities and consolidations in a bilateral, symmetrical or sub pleural distribution. It’s a disease with diagnostic difficulty so a multidisciplinary diagnosis was approached for the diagnosis i.e., clinical, radiological and histopathological. The prognosis of this condition is variable; it is a reversible disease with risk of progression. Therefore, early diagnosis and disease stratification are important in this disease. Here we report the case of a three-year-old girl child who presented with atypical clinical and radiological features, but had been diagnosed to have c-NSIP.
Case Report
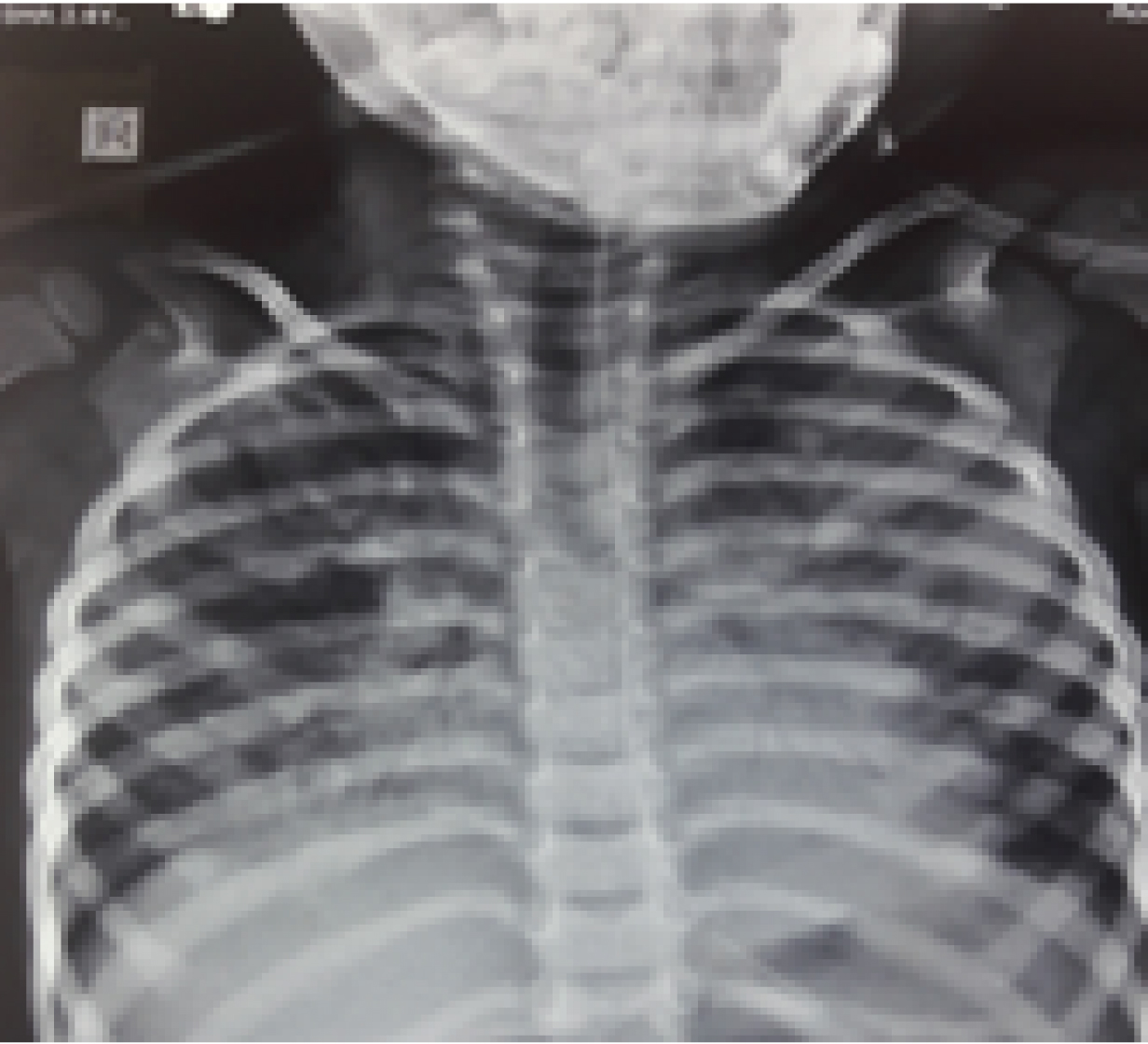
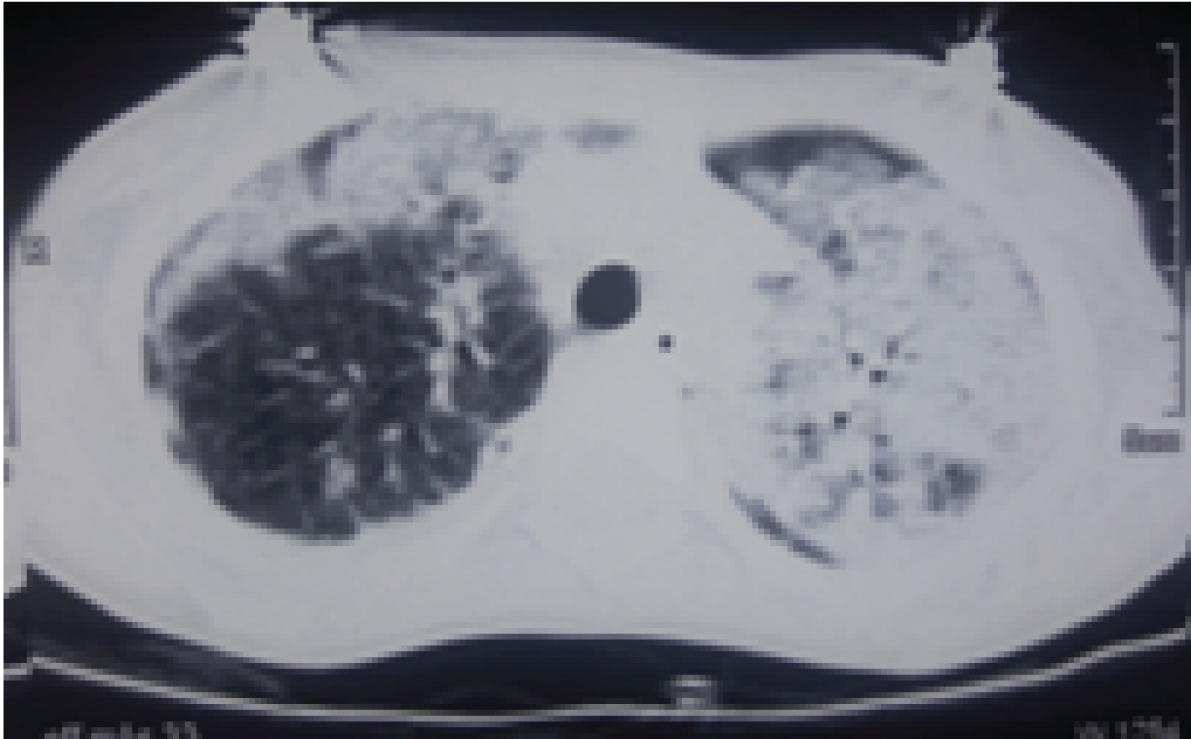
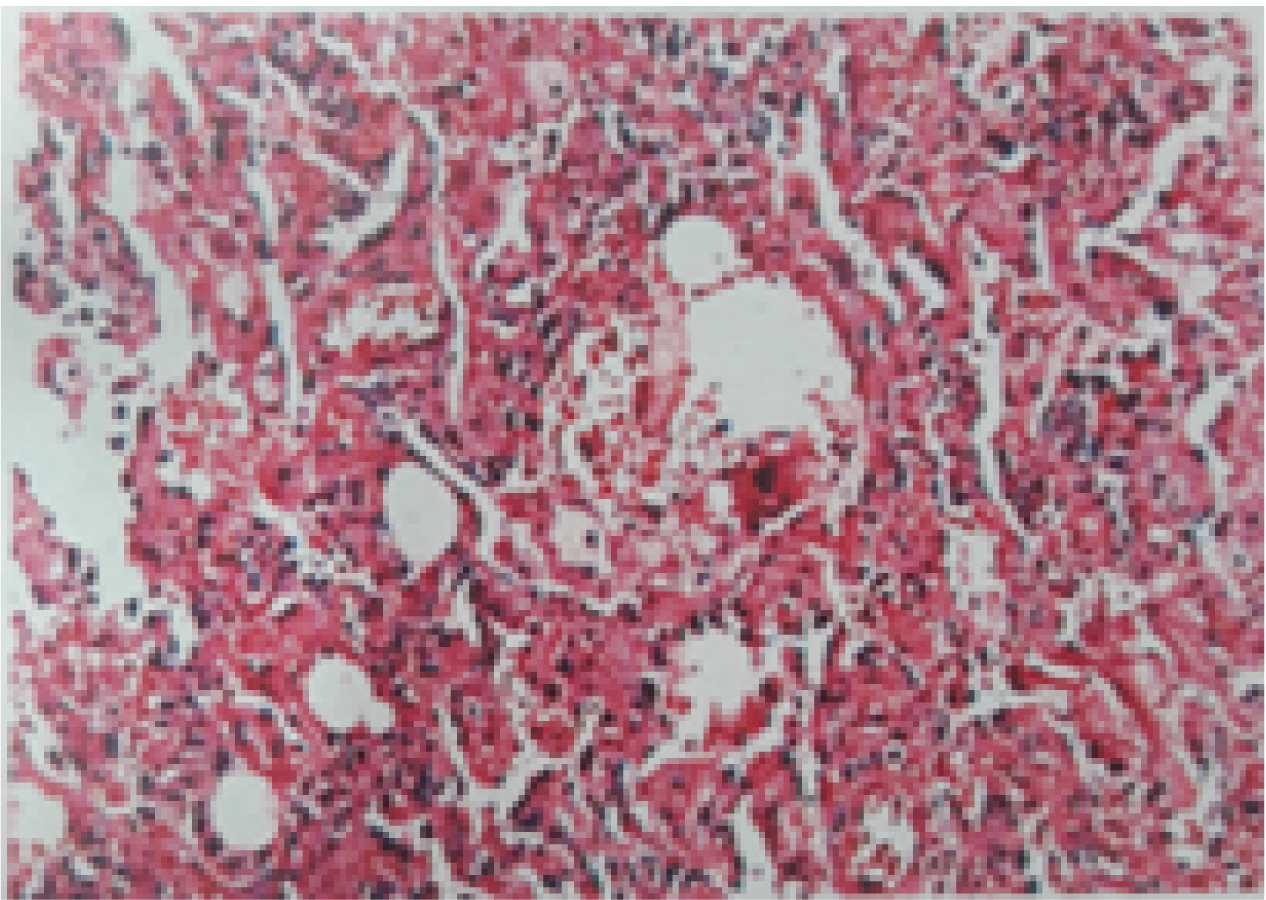
A three-year-old, 2nd born female child to 3rd degree consaguineous marriage was admitted to Paediatric Intensive Care Unit (PICU) with complaints of fever for one month, coughs and hurried breathing for two days. Fever was moderate and intermittent with chills. Cough was productive. She had history of two previous hospitalisation because of lower respiratory tract infections. There was no significant antenatal or post natal history. She had normal development and she was immunised upto her age according to national immunisation schedule. There was a significant family history of elder female sibling death at 4½ years due to recurrent pneumonia, started from the age of one year. On examination, she was afebrile, tachypnoeic moderately malnourished with severe lower chest retractions and bilateral bronchial breathing. Oxygen saturation was 70% on room air and 96% on 3 liter oxygen per minute. There was no clubbing or cyanosis. Child was treated as pneumonia and started on intravenous antibiotics. Laboratory investigations showed lymphocytic leukocytosis [Table/Fig-1]. Chest X-ray showed bilateral patchy opacities and these opacities were more on the left side, as shown in [Table/Fig-2] as multilobar consolidation. Sputum examination was negative for mycobacteria on acid fast bacilli staining by modified Ziehl-Neelsen technique. Sputum culture yielded non candida albicans. High resolution computed tomography of thorax showed linear airspace opacification of both the lungs with air bronchogram with relative sparing of the apical segment of the right upper lobe, lateral segments of right and left lower lobes which were suggestive of lobar and multifocal consolidation [Table/Fig-3]. As the above findings were suggestive of infective aetiology, the patient was not improving with antibiotics and cultures were negative for bacteria, differential diagnoses considered were interstitial pneumonia, sarcoidosis, hypersensitivity pneumonitis, congenital malformations of lung and immunodeficiency. Serum CD3, CD4, CD8 and immunoglobulin levels were done to look for evidence of immunodeficiency, but the results were within normal limits provisional diagnosis of Interstitial Lung Disease (ILD) was made. To rule out the causes of ILD like connective tissue disorders, sarcoidosis, idiopathic interstitial pneumonia, cryptogenic organising pneumonia and langerhans cell histiocytosis, broncho alveolar lavage was done. The results were not suggestive of any typical pattern of any particular diseases like sarcoidosis or hypersensitivity pneumonitis as stated above. Broncho Alveolar Lavage (BAL) fluid culture was sterile for any identifiable pathogenic bacteria. Lung biopsy was done which showed features of non specific interstitial pneumonia with cellular pattern [Table/Fig-4]. Treatment was started with Prednisolone and the child responded initially, but after one month of starting prednisolone, her condition got worsened and she died due to respiratory failure.
In this table, we summarize the major blood and BAL fluid studies done for our patient.
| Parameters | Case | normal range |
|---|
| Haemoglobin (gm/dL) | 12.4 | 11-14 |
| Total count (/mcL) | 12000 | 4000-11000 |
| Differential count | P41L56 | P60L40 |
| Platelet count (/mcL) | 160000 | 150000-450000 |
| ESR (mm/hour) | 12 | 3-13 |
| CRP | Negative | Qualitative |
| Urea (mg/dL) | 21 | 20-40 |
| Creatinine (mg/dL) | 0.9 | 0.7-1.4 |
| Na (mEq/L) | 134 | 135-145 |
| SGOT(IU/L) | 27 | 5-40 |
| SGPT(IU/L) | 6 | 7-56 |
| ALP (IU/L) | 72 | 44-147 |
| S.albumin(gm/dL) | 3.6 | 3.5-5.5 |
| HIV | Negative | - |
| Rheumatoid factor | Negative | - |
| ACE (U/L) | 34 | 8-53 U/L |
| ANA profile | Negative | - |
| c-ANCA | Negative | <1:20 |
| p-ANCA | Negative | <1:20 |
| IgA (mg/dL) | 110 | 80-350 |
| IgG (mg/dL) | 954 | 620-1400 |
| IgM (mg/dL) | 165 | 45-250 |
| CD3+ (n x 106 /L) | 2580 | (1578–3707) |
| CD4+ (n x 106 /L) | 1448 | (870-2144) |
| CD8+ (n x 106 /L) | 804 | (472-1107) |
| BAL-(x 104 cells/mL)(Broncho alveolar lavage) | | |
| Alveolar macrophage | 84 | |
| Neutrophils | 12.5 | |
| Lymphocytes | 0.9 | |
| Eosinophils | 0.2 | |
X-ray showing multilobar consolidation.
CT photograph revealing air space opacification of both the lungs- lobar and mulifocal consolidation.
Histopathology showing cellular pattern of non specific interstitial pneumonia.
Discussion
Interstitial Lung Diseases are a group of diffuse infiltrative lung disorders which are typically characterised by the presence of inflammation and altered lung interstitium [1]. When compared to adults, number of studies conducted are very less in the paediatric age group [2]. Only few studies are conducted in India [3]. Our case had atypical clinical features for NSIP like absence of clubbing at presentation and absence of late inspiratory crackles. Also, High Resolution Computed Tomography (HRCT) features of our case differ from typical radiological features of cellular NSIP. Our case had lobar and multilobar consolidation which is not typical feature of NSIP [4]. Ground glass opacities, which are commonly seen in most cases of NSIP, were absent in our case [4].
One study was conducted in China, which describes clinical, radiological and pathological diagnosis of 25 cases of diffuse parenchymal lung disease in children [5]. Out of these, the most common pattern was non specific interstitial pneumonia (n=9). Five out of these nine patients had cellular variant of non specific interstitial pneumonia, four had mixed pattern and none had fibrotic variant. Our patient also had cellular variant of NSIP. Out of these nine patients with NSIP, only four patients had clubbing. Our patient did not show clubbing.
The key radiological features of NSIP include symmetrical distribution of infiltrates with predominant lower lobe distribution and sub pleural sparing. Ground glass opacity is said to be one of the most important salient feature of NSIP [4]. But our patient’s radiology, findings were asymmetrical although bilateral and with predominant lower lobe involvement. Ground glass opacities were absent in our case, which is very rare. Consolidation is not a typical feature of NSIP although, it is described in some cases [4]. Our patent’s HRCT showed features of consolidation. Also, the characteristic late inspiratory crackles of interstitial lung disease were absent in our case. This may be due to the absence of fibrous change in the lung parenchyma, which was confirmed by the pathologic assessment of Video-Assisted Thoracoscopic Surgery (VATS): biopsied specimen. Saraya T et al., described a case of cellular NSIP that presented With atypical clinical and radiological features of congestive cardiac failure and showed absence of late inspiratory crackles [6].
Interstitial lung diseases are characterised by inflammatory and fibrotic changes affecting the alveolar walls. They are associated with high morbidity and mortality and most of them tend to be chronic [7,8]. There are many classification systems used to describe various types of interstitial lung diseases affecting children. Alexandra AL et al., proposed seven histopathologically distinct subgroups of idiopathic interstitial pneumonias: idiopathic pulmonary fibrosis, nonspecific interstitial pneumonia, respiratory bronchiolitis–associated interstitial lung disease, respiratory bronchiolitis–associated interstitial lung disease/desquamative interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia (frequency <2%), and Lymphocytic Interstitial Pneumonia (LIP) [8].
NSIP can be seen in so many conditions like connective tissue disorders, reactions to certain medications, HIV, hypersensitivity pneumonitis, infection. Most of the cases are idiopathic in which the cause is unknown. Inhaling chemicals or dust, use of certain chemotherapy drugs, or radiation treatment can also produce features similar to NSIP [9].
The two primary forms of NSIP are cellular and fibrotic. The cellular form is characterised by inflammation of the cells of the interstitium, and in the fibrotic form there is thickening and fibrosis of lung tissue which is irreversible [6]. Due to the thickening and scarring, there will be lowering of lung efficiency, so that oxygen levels in the blood are lower. The symptoms are dry cough, shortness of breath during exertion that become worse over time, difficult or laboured breathing, fatigue and clubbing.
Dyspnoea is due to increased work of breathing secondary to decreased compliance, increased resistance or may be steroid related. In c-NSIP, we can hear normal auscultatory sounds, if there is paucity of fibrous components in the lung parenchyma.
The diagnosis of NSIP is based on many factors like clinical examination, Imaging, and histopathology. In BAL, elevated levels of lymphocytes in the fluid have been detected in more than 50% of patients with NSIP, but not always specific [9].
The common findings include ground glass opacities symmetric and diffuse, bilateral (86%) or basal predominance, sub pleural sparing (relatively specific sign), reticular opacities and irregular linear opacities, subpleural reticulation, thickening of bronchovascular bundles, traction bronchiectasis. The Ground Glass Opacity (GGO) or consolidations are usually distributed in peripheral or subpleural spaces and rare in the central area, as well as lower lobe predominance [6]. In advancement of the disease traction bronchiectasis, consolidation and microcystic honey combing are seen.
In c-NSIP, the histologic features included are mild to moderate interstitial chronic inflammation, and type II pneumocyte hyperplasia in areas of inflammation. In the fibrosing type of NSIP, dense or loose interstitial fibrosis that lacks the temporal heterogeneity pattern is observed. Lost lung architecture and interstitial inflammation are also seen in fibrosing NSIP [10].
In cellular type of NSIP, treatment is mainly oral corticosteroids, such as prednisolone and it responds well to treatment; however, in our case initial response to steroids was observed but later it got worsened. When compared to other types of idiopathic interstitial pneumonias, NSIP has better outcome and better response to corticosteroids [11]. Those who are not responding to corticosteroid therapy may require add on therapy with immune-suppressing drugs. Patients with the fibrotic type of NSIP might require both types of drugs to prevent further irreversible fibrosis [12].
Cellular NSIP has excellent prognosis, with low mortality rate. Adult data shows that the five year mortality of cellular NSIP is less than 10% with median survival more than 10 years, whereas in fibrotic NSIP the five years mortality rate is 10% with median survival 6-8 years [13].
Conclusion
We report a female child, diagnosed with cellular variant of non specific interstitial pneumonia with family history and atypical clinical and radiological features. That should be diagnosed early and disease stratification is important in this disease.
[1]. Meyer KC, Diagnosis and management of interstitial lung diseaseTransl Respir Med 2014 2:410.1186/2213-0802-2-425505696 [Google Scholar] [CrossRef] [PubMed]
[2]. Clement A, Task force on chronic interstitial lung disease in immunocompetent childrenEur Respir J 2004 24(4):686-97.10.1183/09031936.04.0008980315459150 [Google Scholar] [CrossRef] [PubMed]
[3]. Sankar J, Pillai MS, Sankar MJ, Lodha R, Kabra SK, Clinical profile of interstitial lung disease in Indian childrenIndian Pediatr 2013 50(1):127-33.10.1007/s13312-013-0034-z22728620 [Google Scholar] [CrossRef] [PubMed]
[4]. Kligerman SJ, Groshong S, Brown KK, Lynch DA, Nonspecific interstitial pneumonia: radiologic, clinical, and pathologic considerationsRadiographics 2009 29(1):73-87.10.1148/rg.29108509619168837 [Google Scholar] [CrossRef] [PubMed]
[5]. Xu D, Chen Z, Chen H, Huang R, Zhao S, Liu X, Application of clinico-radiologic-pathologic diagnosis of diffuse parenchymal lung diseases in children in ChinaPLoS One 2015 10(1):e011693010.1371/journal.pone.011693025569558 [Google Scholar] [CrossRef] [PubMed]
[6]. Saraya T, Takata S, Fujiwara M, Takei H, Cellular non-specific interstitial pneumonia masquerading as congestive heart failureBritish Medical Journal Case Report 2013 2013:pii:bcr201301050210.1136/bcr-2013-01050224001730 [Google Scholar] [CrossRef] [PubMed]
[7]. Dinwiddie R, Sharief N, Crawford O, Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and IrelandPediatric Pulmonology 2002 34(1):23-29.10.1002/ppul.1012512112793 [Google Scholar] [CrossRef] [PubMed]
[8]. Alexandra AL, Myers JL, Idiopathic pulmonary fibrosis: clinical relevance of pathologic classificationAm J Respir Crit Care Med 1998 157(4 Pt 1):1301-15.10.1164/ajrccm.157.4.97070399563754 [Google Scholar] [CrossRef] [PubMed]
[9]. Leslie KO, My approach to interstitial lung disease using clinical, radiological and histopathological patternsJ Clin Pathol 2009 62(5):387-401.10.1136/jcp.2008.05978219398592 [Google Scholar] [CrossRef] [PubMed]
[10]. Du Bois R, King TE Jr, Challenges in pulmonary fibrosis x5: the NSIP/UIP debateThorax 2007 62(11):1008-12.10.1136/thx.2004.03103917965079 [Google Scholar] [CrossRef] [PubMed]
[11]. Kim DS, Collard HR, King TE Jr, Classification and natural history of the idiopathic interstitial pneumoniasProc Am Thorac Soc 2006 3(4):285-92.10.1513/pats.200601-005TK16738191 [Google Scholar] [CrossRef] [PubMed]
[12]. Kondoh Y, Taniguchi H, Yokoi T, Nishiyama O, Ohishi T, Kato T, Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumoniaEur Respir J 2005 25(3):528-33.10.1183/09031936.05.0007100415738299 [Google Scholar] [CrossRef] [PubMed]
[13]. King TE Jr, Clinical advances in the diagnosis and therapy of the interstitial lung diseasesAm J Respir Crit Care Med 2005 172(3):268-79.10.1164/rccm.200503-483OE15879420 [Google Scholar] [CrossRef] [PubMed]