Respiratory tract infections have become increasingly difficult to treat owing to the increase in resistant Gram positive and Gram negative bacteria. Penicillins, fluoroquinolones like levofloxacin, moxifloxacin and macrolides are generally used to treat respiratory tract infections. However, there is an increased concern about antibiotic resistance especially with penicillins and macrolides or azithromycin when used in empirical settings for the management of community acquired upper or lower RTIs caused by Streptococcus pneumoniae or atypical pathogens which are common pathogens responsible for RTI [1].
Garenoxacin mesylate is a novel quinolone with structural modifications favouring a unique pharmacokinetic profile, broad spectrum of activity including resistant strains at extremely low minimum inhibitory concentrations and mutant prevention concentrations indicated in treatment of RTIs. The clinical efficacy and safety of garenoxacin mesylate has been evaluated in phase III clinical studies in subjects with community acquired pneumonia and upper respiratory infections [2-7]. As per the available published literature, the safety profile of garenoxacin mesylate has been well documented in several studies especially in the Japanese population since its marketing approval in 2007 [3-7]. Garenoxacin was approved in India in the year 2013 for treatment of bacterial RTIs. As a regulatory requirement for understanding the safety concerns and clinical utility of garenoxacin in real-world clinical setting in Indian patients with bacterial RTIs, we conducted a post marketing surveillance study.
Materials and Methods
This phase IV study was designed as a single-arm, uncontrolled, open-label, multicenter, observational study to collect the safety information regarding garenoxacin mesylate in treatment of bacterial RTIs across eight established medical centers (2 centers at Gurgaon, 2 at Pune, 1 at Hyderabad, 1 at Srikakulaam, 1 at Vijaywada and 1 at Kolkata) in India and in real life clinical settings. This study was conducted in the period from 2015 to 2017. The duration of treatment was dependent on the severity of the disease with a maximum duration of 14 days. The duration of study was 2 years which involved the process of screening, recruitment, data collection, analysis and assessment report generation. The clinical trial protocol, informed consent form were reviewed and approved by Ethics Committee in accordance with Good Clinical Practices (GCP) guidelines as per International Conference of Harmonization (ICH-E6), World Medical Association Declaration of Helsinki (October 2013), Central Drugs Standard Control Organization (CDSCO) and according to the Indian Council of Medical Research (ICMR) guidelines for biomedical research schedule Y. The study was conducted as per the standard operating procedures of the sponsor and designee to ensure adherence to GCP guidelines.
Inclusion criteria were patients of either sex between the age group of 18 and 65 years with clinical signs and symptoms of RTI with bacteriological aetiology suspected by the investigator during evaluation in real life clinical setting, have provided written informed consent and those willing to comply with all aspects of the study protocol.
Patients excluded were those requiring hospitalisation, with known hypersensitivity to fluoroquinolones or any constituents of the formulation, have received antimicrobial therapy within seven days prior to enrolment, pneumonia acquired in hospital within the past 14 days onset of symptoms, severe complications of RTI which required hospitalisation during past 14 days, any clinically unstable medical condition, known immunocompromised conditions or on anti-tubercular therapy, febrile neutropenia, thrombocytopenia, clinically significant derangements in ALT, AST, bilirubin or any other laboratory parameters, with history of central nervous system disorders such as convulsions, anxiety, confusion, depression and insomnia, with history of cardiac arrhythmias or having risk factors for development of fatal cardiac arrhythmias due to QT prolongation i.e., Torsade de Pointes and pregnant and/or lactating women.
On screening, patients satisfying the inclusion and exclusion criteria were enrolled in the study. Subjects were provided necessary details about the study including the details of the study medication and informed valid consent was obtained before the start of recruitment process. Subjects were screened for medical history, vital signs and physical examination, clinical symptom evaluation and investigations such as complete blood count, alanine transaminase, aspartate aminotransferase, bilirubin, random blood glucose, pregnancy test in females of child bearing age, X-ray chest, Mantoux test and Electrocardiogram (ECG). The primary endpoint of the study was safety assessment in the form of recording the adverse events occurring while on treatment with garenoxacin. The secondary endpoint was assessment of clinical utility in the form of improvement in symptoms after receiving garenoxacin.
Subjects were prescribed a single daily oral dose of garenoxacin mesylate: 2×200 mg Tablets QD (400 mg) for a minimum of 5 days and a maximum of 14 days. The duration of treatment was dependent upon severity of the underlying condition as judged by the investigator. Safety was assessed by evaluating clinical and laboratory parameters. All enrolled subjects were followed up on day 4±1 after the last dose of study drug. The subsequent follow up was done either by a telephone call or by scheduling a clinic visit. All Treatment Emergent Adverse Events (TEAEs) that occurred during the treatment until fourth day after the intake of last dose of the study drug were recorded. The end of the study was the date of the last study visit for the last subject in the study. Safety of garenoxacin was evaluated on the parameters such as changes in the vital signs, laboratory safety parameters or investigations and incidence and rates of adverse events with use of garenoxacin from baseline to the end of study.
Statistical Analysis
A Statistical Analysis Plan (SAP) was finalised to provide details of the analysis. All data was summarised with descriptive statistics for continuous endpoints, and frequency and percentage for categorical endpoints. Sample size was calculated based on the binomial distribution taking into consideration 99% probability of detecting at least one adverse event with an underlying incidence of at least 1%. SAS consisted of all subjects who received at least one dose of garenoxacin and included for analysis. The values at the baseline were compared with End of Treatment (EOT) values using paired t-test. The causality relationship of adverse events with the study drug was determined using WHO scale. For analysis of clinical utility, the incidence of patients with symptomatic improvement were recorded during follow up visits.
To ensure the completeness and accuracy of case report forms, the study was monitored by a designated clinical trial monitor.
Results
The study enrolled 461 subjects of which 97.8% completed the study as per protocol. A total of 461 patients were taken into consideration in SAS because they received at least one dose of garenoxacin. Four subjects withdrew consent, while four subject missed the EOT visit, one was withdrawn due to non compliance with study procedures and one subject was lost to follow up. Total number of subjects at baseline was 461. On follow up, 452 patients visited on visit 1, 25 patients on visit 2 and only 1 on visit 3. The Visit 4 was completed for 451 subjects of which 158 visited the clinic and 293 were telephonically contacted. The adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA).
The study had 438 subjects who received the study drug for five days, 22 subjects received for 10 days and one patient received the study drug for 14 days. The baseline demographic details of the patients are described in [Table/Fig-1]. All the subjects were of Asian origin. Of these enrolled subjects 446 (96.7%) had Upper Respiratory Tract Infection (URTI) and 15 (3.2%) had Lower Respiratory Tract Infection (LRTI). Patient with URTI received garenoxacin mesylate 200 mg two tablets per day for 5 days; whereas patients with LRTI received same therapy for 14 days.
Baseline demographics for Safety Analysis Set (SAS) patients
| Parameters | SAS (N=461 patients) |
|---|
| Age (Mean±S.D.) (years) | 33.7±11.30 |
| Age wise distribution (years) | N (%) |
| 18-30 | 229 (49.7%) |
| 31-40 | 121 (26.2%) |
| 41-50 | 69 (15%) |
| 51-65 | 39 (8.46%) |
| >65 | 3 (0.65%) |
| Gender |
| Male | 268 (58.1%) |
| Female | 193 (41.9%) |
The urine pregnancy test was negative for all the subjects tested. The evaluation of data from all enrolled subjects showed that 211 (45.8 %) subjects had at least one relevant medical/surgical history, with majority of them having medical/surgical history of respiratory system disorders (86.3%). Overall, 98% of the subjects included in this study consumed at least one concomitant medication. Nearly 60% of the subjects consumed paracetamol. This was followed by subjects who consumed Pantoprazole (26.03%) and Cetirizine (approximately 23%).
Incidence and Rate of TEAEs
During the study, 54 TEAEs were reported in 53 (11.5%) subjects as shown in [Table/Fig-2]. No serious TEAE was reported during the study and all the TEAEs were of mild severity. Of the TEAEs reported, no action was taken in 92.6% cases, while in 7.41% cases medication was administered. All the TEAEs were completely resolved. Only 74.1% of the TEAEs were related to study drug as established from the known safety profile of garenoxacin.
Number of patients with system-wise treatment emergent adverse events.
*Skin and subcutaneous tissues
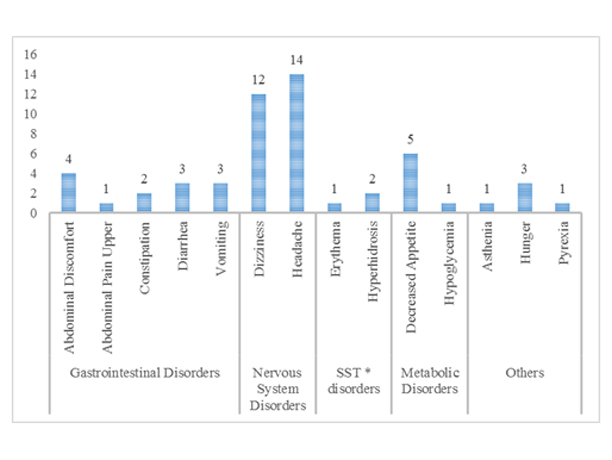
The subjects who experienced TEAEs (11.5%), majority of them experienced TEAEs from nervous system disorders (5.64%) and gastrointestinal disorders (2.82%). The system wise analysis of adverse events is shown in [Table/Fig-2]. The most frequently experienced TEAEs were headache (3.04%), dizziness (2.60%), decreased appetite (1.08%), and abdominal discomfort (0.87%). The study had 13 TEAEs reported from gastrointestinal disorders and six of them were classified as related to the study drug while seven were classified as not related to study drug. Similarly of the 27 TEAEs in nervous system disorders, 21 were related and six were not related to study drug. Five TEAEs reported in general disorders and administration site conditions, four were related to study drug; while all the TEAEs from skin and subcutaneous tissue disorders (3; 0.56%) and metabolism and nutrition disorders (6; 1.30%) were related to study drug. All the TEAEs of upper abdominal pain, asthenia, hunger, hyperhidrosis, erythema, hypoglycaemia, and decreased appetite were related to study drug. No subject experienced any adverse events leading to discontinuation or adverse event of special interest during the study. No subject died, experienced Severe Adverse Events (SAEs), or discontinued from study or treatment because of adverse events.
Changes in the Laboratory Safety Parameters from Baseline to End of Treatment
The analysis of percentage of subjects with ‘out of range laboratory values at baseline vs. EOT showed marked improvement in eosinophil count (52.1% vs 19.5%) and neutrophil count (12.8% vs. 2.22%); lesser improvement in WBC count (12.6% vs. 8.43%), lymphocyte count (20.6% vs. 13.5%), and SGOT (18.7% vs. 15.1%), and minor improvement in RBC count (16.1% vs. 15.5%), platelet count (5.86% vs. 5.32%), monocyte count (1.74% vs. 1.55%), basophil count (13.0% vs. 12.6%), random blood glucose (4.34% vs. 3.55%), SGPT (21.5% vs. 20.2%), direct bilirubin (6.07% vs. 5.76%), and indirect bilirubin (9.11% vs. 8.87%); while it showed minor worsening in haemoglobin (28.2% vs. 29.0%) and total bilirubin (11.3% vs. 14.0%), which was not clinically significant.
Changes in Vital Signs and Electrocardiogram from Baseline to EOT Analysis
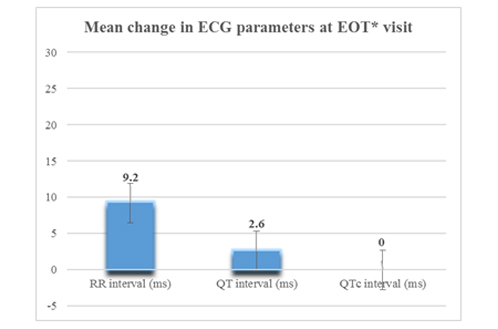
The mean±SD change from baseline to EOT in systolic BP was 0.7±6.99 mmHg, in diastolic BP was −1.1±8.22 mmHg, in pulse rate was −2.5±7.27 bpm, in body temperature was −1.3±1.33 °F, in respiratory rate was −0.4±1.47/minutes, and in weight was 0.1±0.41 kg. The mean ECG parameters for study subjects were stable at all the study visits for all factors including QTc prolongation. The mean change in RR interval, QT and QTc prolongation at the EOT is shown in the [Table/Fig-3]. The mean QTc values at the baseline and at the EOT were 13 millisecond. Only one patient followed up at third visit and his heart rate was 50 bpm.
Changes in Electrocardiogram parameter (ms–millisecond) at the end of treatment.
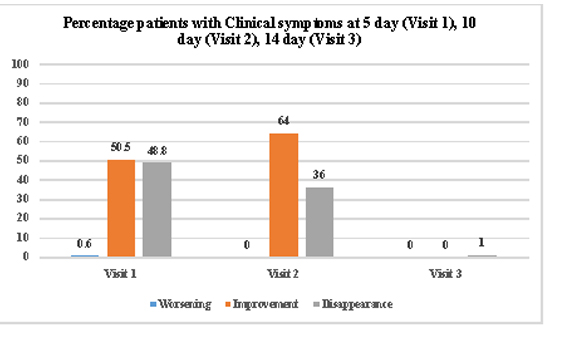
The evaluation of clinical symptoms showed that at EOT visit none of the subject had any persistent clinical symptom assessed during follow up visits as shown in the [Table/Fig-4].
Percentage of patients with clinical symptoms at each visit.
Discussion
Garenoxacin is a broad spectrum novel antibiotic which is very useful in treating severe RTIs and other pathogenic infections. The drug is currently marketed in Japan. In India, garenoxacin has been approved in 2013 for treating bacterial RTIs. Its clinical efficacy has been well proven in clinical studies where it has shown a higher efficacy than the comparator arms. According to the study by Ito M et al., garenoxacin 400 mg/day when administered in patients with pharyngolaryngitis, tonsillitis, otitis media and sinusitis demonstrated 98% complete eradication rate [2]. According to Kohno S et al., there was non-significant difference in efficacy rates between garenoxacin 400 mg once a day (94.9%) and levofloxacin 100 mg three times a day (92.8%) while treating patient with bacterial pneumonia [3]. There was a statistical difference in the eradication rates with garenoxacin (100%) and levofloxacin (87.8%) in the same patients. According to study conducted by Kobyashi H et al., and Takagi H et al., the clinical efficacy rates were 89% and 100% against Penicillin Resistant Streptococcus pneumoniae (PRSP) and Quinolone Resistant Streptococcus pneumoniae (QRSP) pathogens [4,5]. Similarly, the molecule was suggested to have a superior safety profile with negligible incidence of gastrointestinal, central nervous system and cardiovascular side effects including arrhythmias arising as a result of QTc prolongation. This study was therefore conducted to further establish its clinical safety in Indian population post approval by the regulatory authorities. The study was designed to assess the safety profile while assessing the clinical utility of garenoxacin mesylate in real world of Indian outpatient community settings, especially in patients with bacterial RTIs.
The major outcome of study included evaluation of TEAEs in management of community acquired RTIs in Indian population. In a Japanese study by Hori S et al., conducted in 6412 patients, the rate of incidence of TEAEs was 3.45% (221/6, 412 patients) that included gastrointestinal disorders (0.87%, 56 patients) such as diarrhoea [6]. In post marketing surveillance clinical studies conducted by Izumikawa K et al., the adverse drug reactions observed were mainly gastrointestinal disorders (2.9%), infection and infestation (1%), nervous system disorders (1%) and skin and subcutaneous tissue disorder (1%) [7,8]. In the current study, 54 TEAEs were reported in 11.5% (53/461) subjects. One patient had developed two adverse events. Majority of TEAEs in our study were of nervous system disorders (5.64%) and gastrointestinal disorders (2.82%), with most frequently experienced TEAEs being headache (3.04%), dizziness (2.60%), decreased appetite (1.08%), and abdominal discomfort (0.87%). No SAE or death was reported in the current study as compared to the Hori S et al., study that reported 13 SAEs (0.19%, 12 patients) [6].
Gastrointestinal adverse events are commonly observed with use of antibiotics, commonly with penicillins. In a meta-analysis conducted by Gillies M et al., diarrhoea was the most common gastrointestinal disorder reported with penicillins including amoxicillin/clavulanic acid (88/503 patients) with documented Odds Ratio (OR) of 3.3 that was statistically significant (p<0.001) [9]. In the current study, the most common gastrointestinal disorder reported was abdominal discomfort (4, 0.8%) with only few patients (3, 0.6%) reporting diarrhoea with the use of garenoxacin mesylate.
Incidence of neurological disorders reported in pharmacovigilance study by Oreagba IA et al., was 22.4% (121 patients), of which 48.7% (59 patients) reported with use of levofloxacin, 36.3% (44 patients) with ciprofloxacin and 11.5% (14 patients) with ofloxacin. Headache, insomnia and dizziness were the commonly reported neurological adverse effects with incidence of 4.3%, 4.1% and 3.3% (23, 22 and 18 patients) respectively. Insomnia was more commonly reported neurological adverse effect with levofloxacin [10]. In the current study, the incidence of nervous system related adverse events with garenoxacin mesylate was 5.64% (26 patients), of which commonly observed adverse events were headache (14 patients, 3.04%) and dizziness (12 patients, 2.6%).
Analysis of spontaneous adverse drug reports conducted by Tailor SAN et al., revealed 27 reports of metabolic and nutritional related adverse events with use of gatifloxacin. Of these reports, 93% (25 reports) were documented to be either due to hypoglycaemia or hyperglycaemia. Adverse events related to glucose metabolism i.e., either hypoglycaemia or hyperglycaemia were also reported with levofloxacin and moxifloxacin (11% and 10% respectively) [11]. In the current study, 1.3% TEAEs (6 patients) were related to metabolism and nutritional disorders. Of these TEAEs, decreased appetite was observed in 1.08% (5 patients) and only one patient had hypoglycaemia with use of garenoxacin mesylate.
In the Hori S et al., study ECG measurements were performed in 286 patients and 2.8% (8/286 patients) were observed with abnormal values [6]. Risk of cardiovascular adverse events remains a concern with use of antibiotics especially with macrolides and fluoroquinolones. In a study conducted by Ray WA et al., there was a small absolute increase in cardiovascular deaths with five days of azithromycin therapy. When compared with amoxicillin, there were 47 additional cardiovascular deaths per one million courses of azithromycin therapy. This cumulative effect of azithromycin was attributed to its proarrhythmic potential [12]. Study conducted by Haverkamp W et al., reported treatment emergent cardiac events in 698 patients (6.6%) with use of oral moxifloxacin. A total of 14 patients who were administered moxifloxacin developed ventricular arrhythmia, ventricular fibrillation or ventricular tachycardia. Sixteen patients treated with moxifloxacin reported cardiac arrest, of which six had cardiac comorbidity. After moxifloxacin treatment, an incremental prolongation in QTc interval of 6.4 ms compared to 0.6 ms with comparator quinolone treated arm was observed [13]. In the current study too, there was no change noted in QTc interval calculated at the EOT visit for patients with URTI or LRTI who were administered garenoxacin for 5 to 14 days.
It was observed that all the TEAEs encountered were of mild intensity. No action was required for 50 (92.6%) subjects; whereas medication was prescribed to provide symptomatic relief in only 4 (7.4%) cases and all TEAEs were completely resolved at the end of the study.
Again as highlighted by Hori S et al., and Izumikawa K et al., the clinical assessment of the patients showed that at EOT visit none of the subject had any persistent clinical symptom that required discontinuation or treatment with alternative drugs or antibiotics [6-8].
There was no new adverse event experienced in the current study which warranted any changes in the prescribing information. The current findings of the safety analysis are in accordance to the available safety database.
Limitation
Limitation of the study included loss of patients during follow up and clinical utility should have included radiological and bacteriological improvement.
Conclusion
Garenoxacin mesylate represents a novel generation fluoroquinolone offering broad spectrum action. The current study establishes an excellent tolerability and safety profile for garenoxacin mesylate in Indian population and promises complimentary prudent strategy for the management of community acquired RTIs especially in Indian outpatient settings.
Disclosure
Dr. B L N Prasad, Dr. D N Hambire, Dr. Penurkar M, Dr. Praveen, Dr. Pasha A, Dr. Garg H, Dr. Gupta N, and Dr. Dhar R had no conflict of interest with Majesta, a division of Glenmark Pharmaceuticals Ltd that supported the conduct of this clinical study. Details of eight sites are as follows:
(1) Dr. Himanshu Garg. Artemis Health Institute, Sector-51, Gurgoan, 122001. (2) Dr. Neeraj Gupta. Paras Hospitals, C-1, Sector-43, Sushant Lok, Gurgaon-122002. (3) Dr. Dattatraya Hambire/Dr. Reema Kashiva. Noble Hospitals, 153, Magarpatta City Road, Hadapsar, Pune-411013. (4) Dr. Mukund Penurkar. Sanjeevan Hospital, Plot No. 23 Off Karve Rd, Erandwane, Pune–411004. (5) Dr. Athar Pasha. Care Hospital, Road No. 10, Banjara Hills, Hyderabad-500034. (6) Dr. BLN Prasad. Rajiv Gandhi Institute of Medical Sciences and Government General Hospital, Srikakulam-532001. (7) Dr. Maddirala Praveen. Praveen Cardiac Center, Praveen Hospitals, 32-9-18, Moghalrajapuram, Madhu Garden Center, Vijayawada-520010. (8) Dr. Raja Dhar. Fortis Hospital, 730 Anandapur, Kolkata-700107.