ALK-1 Positive Anaplastic Large Cell Lymphoma Presenting as Extensive and Exclusive Osseous Involvement: Report of a Rare Association and Review of Literature
Swetha Lakshmi Narla1, Ann Joseph Kurian2, Annapurneswari Subramanyan3, Ashok Parameswaran4
1 Junior Consultant, Department of Histopathology, Apollo Cancer Institute, Chennai, Tamil Nadu, India.
2 Senior Consultant, Department of Histopathology, Apollo Cancer Institute, Chennai, Tamil Nadu, India.
3 Senior Consultant and Head, Department of Histopathology, Apollo Cancer Institute, Chennai, Tamil Nadu, India.
4 Chief of Laboratory Service, Department of Histopathology, Apollo Cancer Institute, Chennai, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Swetha Lakshmi Narla, 320 Anna Salai, Teynampet, Chennai-600035, Tamil Nadu, India.
E-mail: drswetha.gmc2k2@gmail.com
Primary Bone Lymphomas (PBLs) are uncommon. Most of these tumours are Non Hodgkin Lymphoma (NHL) with Diffuse Large B-cell Lymphoma (DLBCL) being the most common. Anaplastic Large Cell Lymphoma (ALCL) constitutes approximately 2% of all NHL and involves both nodal and extra nodal sites. ALCL presenting as exclusive and extensive bone involvement is rare. We report a case of 31-year-old man who presented with osseous involvement only. Imaging showed multiple lytic lesions involving the vertebral column, ribs and sternum. Clinical differential diagnosis included osteomyelitis and bone neoplasms. Histopathology and immunohistochemistry of the lytic lesion proved it to be Anaplastic Lymphoma Kinase (ALK-1) positive ALCL. The patient relapsed three months after completion of six cycles of combination chemotherapy with Cyclophosphamide, Hydroxydaunorubicin, Vincristine and Prednisone (CHOP).
Anaplastic lymphoma kinase, Bone involvement, Non hodgkin lymphoma, Primary bone lymphoma
Case Report
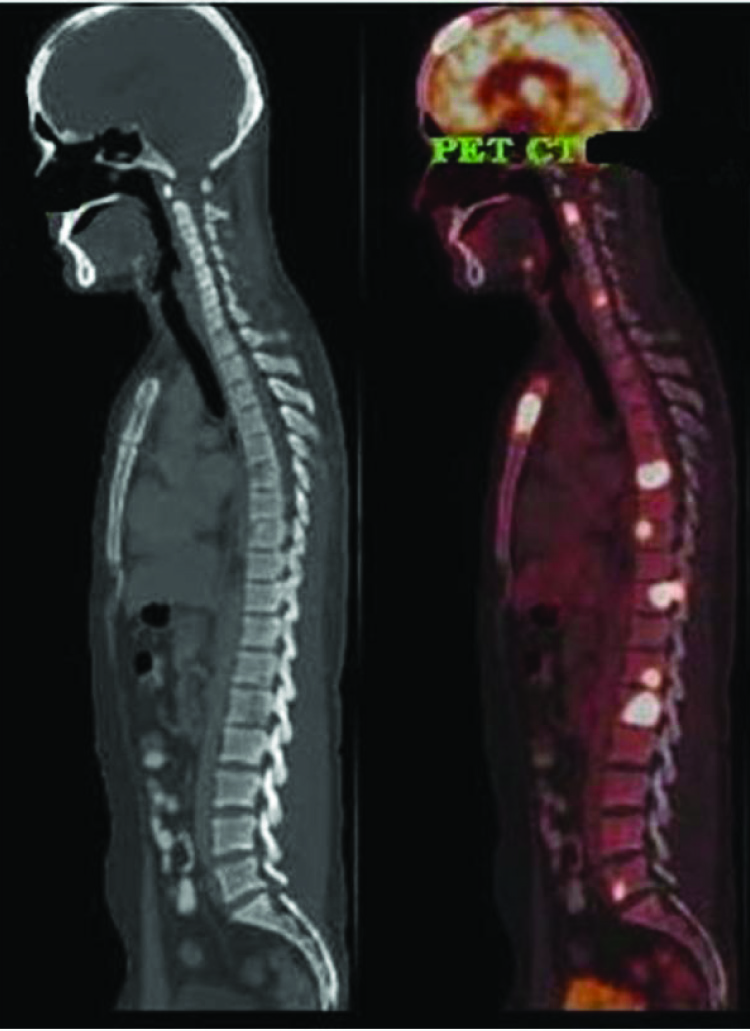
We report a case of 31-year-old man who presented with a history of fever and chest pain of one month duration. Physical examination did not reveal any lymphadenopathy, organomegaly or cutaneous involvement. A complete haemogram, serum uric acid, calcium, phosphorus, alkaline phosphatase, liver and renal function tests were within normal limits. Skin Mantoux test was negative. There was an elevation in Erythrocyte Sedimentation Rate (ESR) (107 mm/hour) and Lactate Dehydrogenase (LDH) (232 U/L). Chest X-ray and Magnetic Resonance Imaging (MRI) showed multiple skeletal lytic lesions. The 18 Fluro Deoxyglucose (FDG), Positron Emission Tomography (PET)/Computed Tomography (CT) scan showed expansile lytic lesions with increased FDG avidity involving D7 to D10 vertebrae, ribs and sternum [Table/Fig-1]. Other investigations for chest pain like Electrocardiogram (ECG) were not done since imaging showed multiple lytic lesions. There was no lymphadenopathy or organomegaly. Based on the extensive bone involvement and complaints of chest pain, a clinical diagnosis of osteomyelitis or bone neoplasms was considered.
An 18F FDG PET/CT scan showing extensive lytic lesions with increased FDG avidity involving multiple bones.
Biopsy of the vertebral lytic lesion revealed sheets of atypical large pleomorphic cells with embryoid like nuclei, prominent nucleoli and frequent mitotic figures on a polymorphous background which was composed of lymphocytes, eosinophils and histiocytes. These atypical cells were seen traversing the bony trabeculae [Table/Fig-2]. A possibility of high grade lymphoma was thought of and proceeded with further workup.
a) ALCL showing large pleomorphic hallmark cells with eccentric nucleus, prominent nucleoli and abundant cytoplasm (H&E, 10X); b) ALCL showing large pleomorphic hallmark cells with eccentric nucleus, prominent nucleoli and abundant cytoplasm (H&E, 40X); c) Immunohistochemistry of ALCL showing expression of CD30 (40X); d) Immunohistochemistry of ALCL showing expression of ALK-1 (40X); e) Immunohistochemistry of ALCL showing expression of CD4 (40X); f) Ki-67 labelling index 15-20% (40X); g) no expression of CD3 (40X); h) CD20 (40X); i) LCA (40X) and j) CD15 (40X).

Histologically, a possibility of lymphoma was suggested. Immunohistochemically, these large cells showed positivity for CD 30 (Dako; Ber-H2), ALK-1 (Dako; Alk1), CD4 (Dako; Polyclonal) and were negative for Cytokeratin (Dako; AE1+AE3), CD3 (Dako; Polyclonal), CD20 (Dako; L26), CD15 (Dako; Carb-3), LMP (Cellmarque; CS 1-4), LCA (Dako; 2b11+pd7/26), CD56 (Dako; Polyclonal), CD2 (Biogenex; AB75), CD5 (Dako; SP19), CD7 (Dako; CBC.37) and CD8 (Dako; Polyclonal). The proliferation index (Ki 67) (Dako; MIB1), was around 15-20% [Table/Fig-2]. Bone marrow aspiration, trephine biopsy and cerebrospinal fluid examination done for staging showed no evidence of lymphomatous involvement. Patient was categorised as clinical stage IV (Ann Arbor staging) according to the sites of involvement. The patient unfortunately relapsed after 3 months of completion of 6 cycles of combination chemotherapy (CHOP). Patient was suggested for genetic analysis to evaluate the probable cause of relapse; however, the patient was lost for follow up.
Discussion
Although, NHLs, both T and B-cell types although, frequently involve bone marrow, they rarely produce bone lesions. PBL is a rare disease, first described by Oberling in 1928 [1], that constitutes around 7% of all malignant bone tumours, 4-5% of all extranodal NHL and <1% of all malignant lymphomas [2-4]. World Health Organisation (WHO) classified bone lymphomas into four groups-Group I: lymphoma with single bone involvement with or without regional lymph node involvement, Group II: lymphoma with multiple bone sites, but no visceral/lymph node involvement, Group III: bone and other multiple visceral or lymph node involvement, Group IV: lymphoma involving any site and found on bone biopsy done to rule out possible involvement [3]. Our patient presented with multifocal bone involvement only and hence, belongs to Group II. Most bone lymphomas are NHL, DLBCL being the most common. There is a slight male predominance with an age range 40 to 60 years [2]. Any part of the skeleton may be involved, but long bones (femur, tibia) are the most common sites of involvement [4]. Glotzbecker MP et al., have reported bone pain, soft tissue swelling or pathological fracture as the most common symptoms [5].
ALCL is a distinct type of NHL that has been recognised as a specific entity under T-cell neoplasms in the new WHO classification of lymphomas. ALCL constitutes 2% of NHL [6] and commonly involves nodal sites. Primary systemic ALCL has increased frequency of bone marrow involvement (30%) and extranodal involvement, including skin (21%), bone (17%), soft tissues (17%), lung (11%), liver (8%), rarely, gastrointestinal tract and central nervous system [6]. ALCL is characterised by pleomorphic large lymphoid cells that express CD30 [7]. It was first described in 1985 and was included in WHO classification of lymphoid neoplasms in 2001. The latest WHO classification categorises ALCL as ALK-1 positive and ALK-1 negative based on ALK-1 protein expression by the tumour cells. There are no morphological differences between the two categories except that the neoplastic cells in ALCL, ALK- are larger and more pleomorphic than those seen in classical ALCL, ALK+. ALK-1 positive cases have a better prognosis than ALK-1 negative cases. The overall five year survival rate in ALCL ALK+ is around 80% compared to 48% in ALCL ALK- cases [4]. Translocation (2;5) involving ALK-1 gene on chromosome 2p23 and Nucleophosmin gene (NPM) on chromosome 5q35 occurs in 20-60% of cases. ALK protein may be detected in most cases (60-70%) of systemic ALCL by immunohistochemistry. Irrespective of whether they express T Cell Receptor (TCR) antigens or not, 90% of ALCL, ALK+ show clonal rearrangement of the TCR genes. While ALCL has been described in nodal and extra nodal sites, bone involvement is rare [8,9]. Few adult and paediatric cases of ALCL with exclusive bone involvement have been described [7,10,11]. Review of the literature by Nagasaka T et al., identified around 16 cases of ALCL presenting with bone lesions. The mean patient age of these patients was 32 years (range, 4 to 63 years) [7]. There was a male predominance with a male/female ratio 3:1. Long bones and spine were the most common sites of involvement. Despite treatment, 10 patients (62.5%) died of disease within two years of diagnosis. Differential diagnosis of ALCL of bone includes eosinophilic granuloma in children, osteomyelitis and malignant small round cell tumours in young adults, which can be excluded with immunohistochemical and molecular studies. Immunophenotypically ALCL cells are positive for CD30, Leukocyte Common Antigen (LCA) and Epithelial Membran Antigen (EMA). More than 60% of the cases express one or more T-cell antigens such as CD3, CD43, etc. In a few cases of ALCL, there is neither T nor B-cell antigen expression and these are designated as “null cell type” [11]. In our case, the tumour cells expressed CD30, ALK1 and CD4, ruling out the possibilities of eosinophilic granuloma, osteomyelitis and other round cell tumours. Gianelli U et al., examined 28 cases of primary bone lymphoma and 26 cases of systemic lymphoma involving bone. About 3 cases were of the null cell phenotype with ALK-1 expression [12]. A subset of ALCLs was found to express cytotoxic proteins like granzyme B. Hence, it was proposed that ALCL originated from lymphocytes with cytotoxic potential. However, this expression did not appear to have any significant impact on prognosis and the clinical and biologic significance of granzyme B in ALCL is unclear. Although, clinical and imaging studies aid in diagnosis, a definitive diagnosis of ALCL is possible only by correlating the findings of histopathology, immunohistochemistry and molecular studies.
Conclusion
ALCL is a well described entity with a varied spectrum of presentations. ALCL with extensive and exclusive bone involvement at initial presentation is rare. A thorough clinical examination and a high index of suspicion are required for diagnosis. Lymphoma involving bone should be included in the differential diagnosis of a young adult presenting with multiple lytic bone lesions. Histopathological and immunophenotypic examination is mandatory for diagnosis.
[1]. Oberling C, Les Reticulosarcomes et les reticuloendothelio sarcomas dela moelle osseuse (sarcomas d’Ewing)Bull Assoc Fr Etude Cancer 1928 17:259-96. [Google Scholar]
[2]. Kitsoulis P, Vlychou M, Papoudou-Bai A, Karatzias G, Charchanti A, Agnantis NJ, Primary lymphoma of boneAnticancer Res 2006 26:325-37. [Google Scholar]
[3]. Delsol G, Falini B, Campo E, Jaffe ES, in World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, Anaplastic large cell lymphoma, ALK-positive, (IARC Press, Lyon, France) 2008 :308-11. [Google Scholar]
[4]. Maruyama D, Watanabe T, Beppu Y, Kobayashi Y, Kim SW, Tanimotok Primary bone lymphoma: A new and detailed characterization of 28 patients in a single Institution studyJpn J Clin Oncol 2007 37:216-23.10.1093/jjco/hym00717472971 [Google Scholar] [CrossRef] [PubMed]
[5]. Glotzbecker MP, Kersun LS, Choi JK, Wills BP, Schoffer AA, Dormans JP, Primary non Hodgkin’s lymphoma of bone in childrenJ Bone Joint Surg 2006 88:583-94.10.2106/JBJS.D.0196716510826 [Google Scholar] [CrossRef] [PubMed]
[6]. Rahmat K, Wastie M, Abdullah B, Primary bone lymphoma: Report of a case with multifocal skeletal involvementBiomed Imaging Interv J 2007 3(4):e5210.2349/biij.3.4.e5221614300 [Google Scholar] [CrossRef] [PubMed]
[7]. Nagasaka T, Nakamura S, Medeiros LJ, Juco J, Lai R, Anaplastic large cell lymphomas presented as bone lesions: a clinicopathologic study of six cases and review of the literatureMod Pathol 2000 13(10):1143-49.10.1038/modpathol.388021111048810 [Google Scholar] [CrossRef] [PubMed]
[8]. Lones MA, Sanger W, Perkins SL, Medeiros LJ, Anaplastic large cell lymphoma arising in bone: report of the monomorphic variant with the t (2,5) (p23; q 35) translocationArch Pathol Lab Med 2000 124:1339-43. [Google Scholar]
[9]. Eyre TA, Khan D, Hall GW, Collins GP, Anaplastic lymphoma kinase-positive anaplastic large cell lymphoma: current and future perspectives in adult and paediatric diseaseEur J Haematol 2014 93:455-68.10.1111/ejh.1236024766435 [Google Scholar] [CrossRef] [PubMed]
[10]. Gajendra S, Sachdev R, Lipi L, Goel S, Misra R, ALK positive anaplastic large cell lymphoma presenting as extensive bone involvementJ Clin Diagn Res 2015 9:XD04-XD05.10.7860/JCDR/2015/11180.543725738071 [Google Scholar] [CrossRef] [PubMed]
[11]. Nayak HK, Nishant R, Sinha NK, Daga MK, Anaplastic large T-cell lymphoma presenting as an isolated osseous involvement: a case report and review of the literatureBMJ Case Rep 2013 2013:pii:bcr201300930810.1136/bcr-2013-00930823605839 [Google Scholar] [CrossRef] [PubMed]
[12]. Gianelli U, Patriarca C, Moro A, Ponzoni M, Giardini R, Massimino M, Lymphomas of the bone: a pathological and clinical study of 54 casesInternational Journal of Surgical Pathology 2002 10:257-66.10.1177/10668969020100040312490975 [Google Scholar] [CrossRef] [PubMed]