Congenital Adrenal Hyperplasia– A Rare Cause of Central Precocious Puberty—A Case Report
Jaydeep Majumdar1, Abhijeet Sharan2, Sarmishtha Mukhopadhyay3, Bhaskar Ghosh4, Sarbani Sengupta5
1 DNB Postgraduate Trainee, Department of General Medicine, B R Singh Hospital and Centre for Medical Education and Research, Sealdah, Kolkata, India.
2 DNB Postgraduate Trainee, Department of General Medicine, B R Singh Hospital and Centre for Medical Education and Research, Sealdah, Kolkata, India.
3 Senior Divisional Medical Officer, Department of General Medicine, B R Singh Hospital and Centre for Medical Education and Research, Sealdah, Kolkata, India.
4 Additional Chief Health Director, Department of General Medicine, B R Singh Hospital and Centre for Medical Education and Research, Sealdah, Kolkata, India.
5 Additional Chief Health Director, Department of General Medicine, B R Singh Hospital and Centre for Medical Education and Research, Sealdah, Kolkata, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Jaydeep Majumdar, House No. 4/24, Viveknagar, Kolkata-700075, India.
E-mail: jaydeepsskm@gmail.com
The term ‘Precocious puberty’ refers to the appearance of pubertal signs at an earlier age than established standard. Precocious puberty may be central or peripheral. Central precocious puberty is caused by early maturation of hypothalamic-pituitary-gonadal axis. Peripheral precocious puberty is due to inappropriate secretion of sex steroid hormones from adrenal gland or gonad or from exogenous source and does not involve hypothalamo-pituitary-gonadal axis. We report a rare case where inspite of clinical and hormonal evidence of central precocious puberty ultimately an adrenal pathology was established in a six-year-old boy.
Adrenal gland hyperplasia, Central precocity, Hypothalamo-pituitary-gonadal axis, Sex steroid
Case Report
A six-year-old boy, born out of non-consanguineous marriage presented with gradually increased phallic size with early morning erection over last 10 months. He also developed pubic and axillary hair over last six months along with masculine change in voice unusual for his age. Interestingly, over last few months he had abrupt height acceleration, approximately 5 cm in last six months, disproportionately greater than his classmates. His behaviour became aggressive recently though his scholastic performance was satisfactory. Enquiry revealed a peculiar salt craving nature of the boy. There was no complaining of seizure or inappropriate episodes of laughter. The parents denied any history of head injury, cranial irradiation, meningitis, encephalitis or androgen exposure of their son. His birth history and perinatal period were uneventful. There was no history of unexplained neonatal death, ambiguous genitalia or precocity in family members.
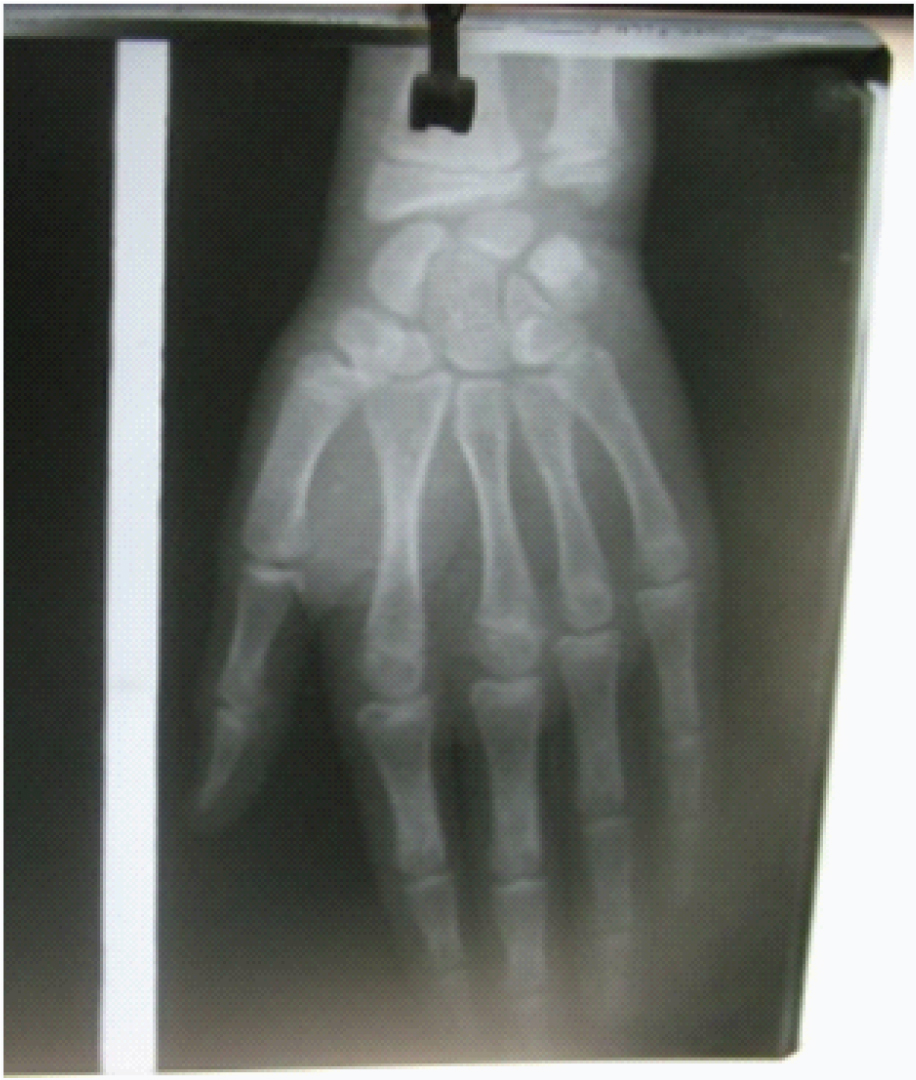
General survey was unremarkable including a blood pressure of 90/60 mmHg. Anthropometric measurement included height- 134 cm (+3.6 SDS), weight- 33 kg (+5.7 SDS) with a sex adjusted mid parental height 165.5 cm (25th percentile). Testicular volume was bilaterally 12 ml, pubic hair was of Tanner stage 3 with presence of axillary hair and stretched penile length was 10 cm [Table/Fig-1]. Height age of the patient was 11.5 years. Bone age of the patient was 9-14 years [Table/Fig-2]. Biochemical investigations and hormonal assay are tabulated in [Table/Fig-3,4], respectively.
External genitalia showing enlarged testes and pubic hair of Tanner stage 3.

X-ray left hand depicting bone age 9-14 years.
Biochemical investigations.
| Parameters | Value in patient | Reference range |
|---|
| Serum sodium | 140 mEq/L | 135-150 mEq/L |
| Serum potassium | 4.1 mEq/L | 3.5-5 mEq/L |
Hormonal assay to evaluate Hypothalamo-Pituitary-Gonadal (HPG) axis.
| Parameters | Value in patient | Reference Range |
|---|
| LH (basal) | 4.02 mIU/ mL | 0.3-6.0 mIU/mL (prepubertal male) |
| LH (40 minutes after injection 100 mcg subcutaneous triptorelin injection) | 29.38 IU/ mL | >0.004-0.005 IU/mL |
| LH (4 hours after injection 100 mcg subcutaneous triptorelin injection) | 28.98 IU/ mL | >0.004-0.005IU/L |
| FSH (Follicle Stimulating Hormone) | 1.40 mIU/ mL | <6.7mIU/mL(0-7 years, male) |
| Serum testosterone | 599 ng/ dL | <7-20 ng/dL (6 months-9 years) |
| FT4 (Free Thyroxine) | 1.01 ng/dL | 0.9-1.7 ng/dL |
| TSH (Thyroid Stimulating Hormone) | 4.36 μIU/mL | 0.5-5.0 μIU/mL |
| Plasma renin | 2.6ng/mL/hour | 0.2-3.3 ng/mL/hour |
| Plasma aldosterone (in supine position) | 30ng/dL | 5-80 ng/dL |
| ß- HCG (Human Chorionic Gonadotropin) | 0.10 mIU/ ml | 0-5 mIU/mL |
| Serum DHEAS (Dehydroepiandrosterone Sulfate) | 1730 μg/ dL | 15- 333 μg/ dL (In males below 11.5 years) |
| 17 hydroxyprogesterone | 4050 ng /dL | 7-170 ng/ dL (3-14 years) |
| 17-hydroxyprogesterone 60 minutes after 250 microgram synacthen | 87000 ng/dL | <1000 ng/dL |
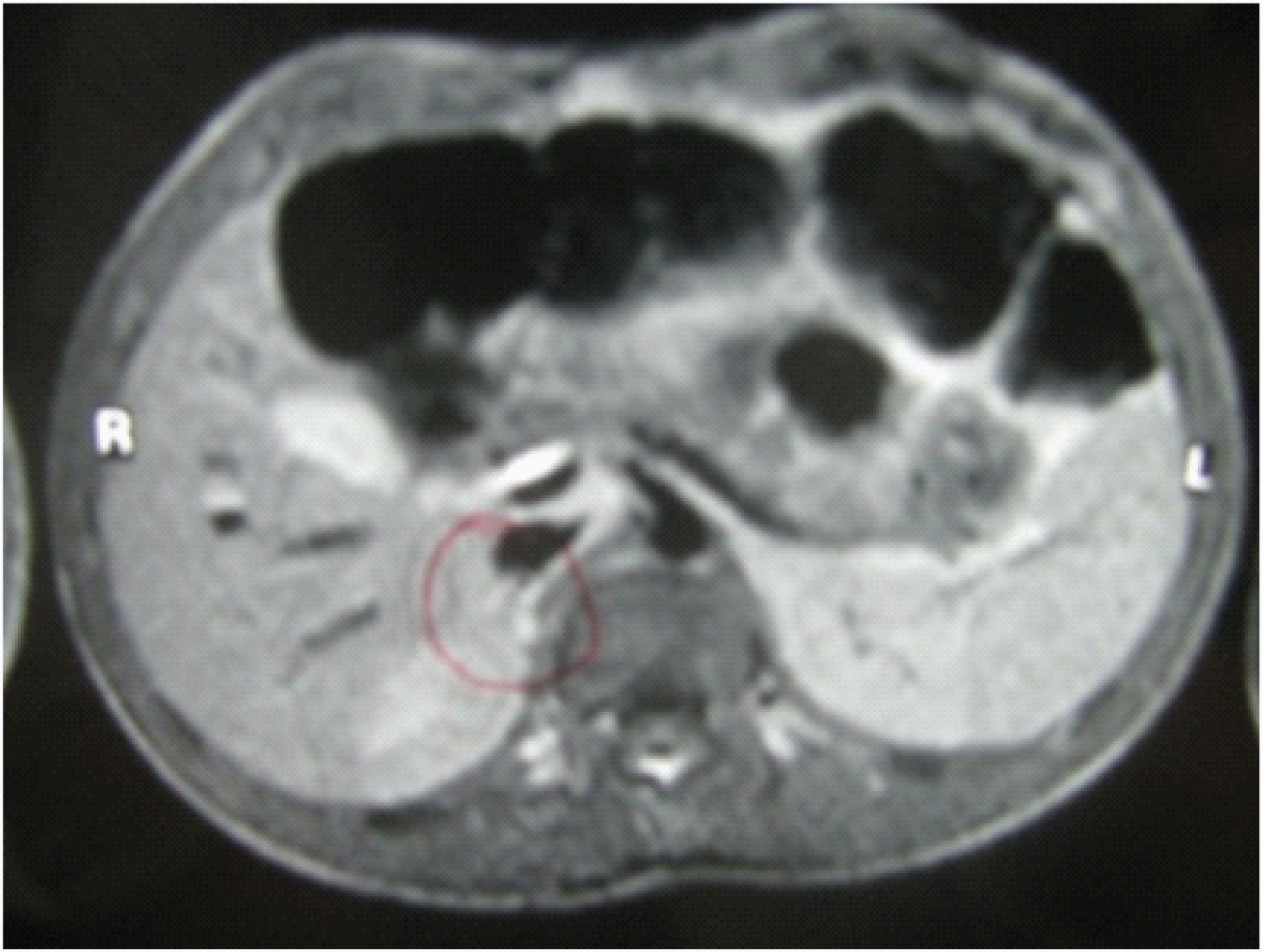
The raised level of serum testosterone indicated precocity whereas raised basal and triptorelin stimulated Luteinizing Hormone (LH) level denoted central precocious puberty. MRI brain with contrast was done which was found to be normal. Contrast enhanced CT scan and MRI abdomen both revealed adrenal hyperplasia, more prominently on the right side [Table/Fig-5,6].
CT scan abdomen showing adrenal hyperplasia on the right side.
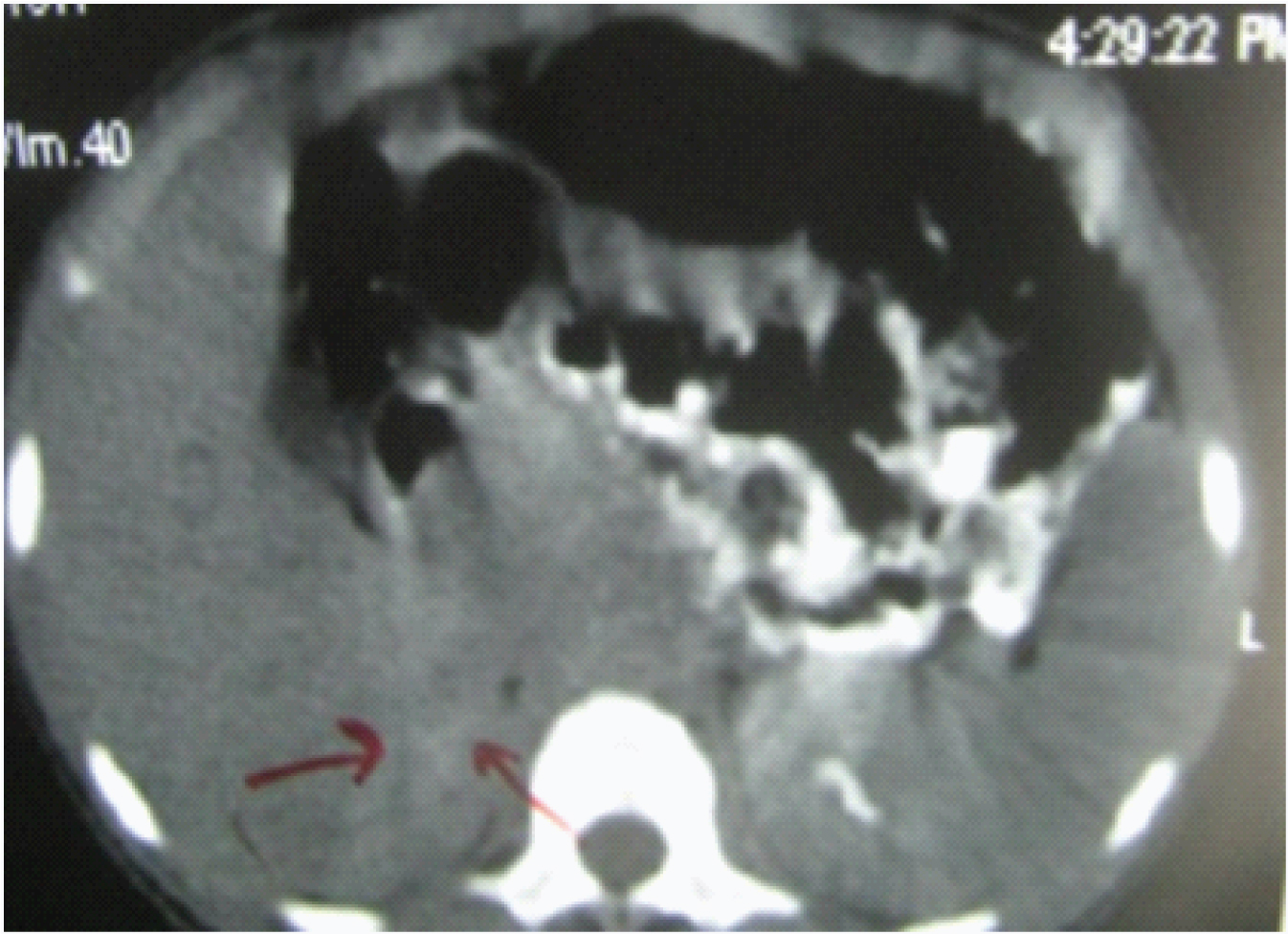
abdomen showing adrenal hyperplasia more prominently on the right side.
A diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency complicated by early HPG axis maturation due to prolonged gonadal steroid exposure producing Central Precocious Puberty (CPP) was confirmed. Treatment was started with tablet hydrocortisone 5 mg thrice daily and the patient responded remarkably. The patient is in regular follow up for last five years and is continuing treatment with tablet hydrocortisone till now. There is no progression of his pubertal development and bone age further. The patient was advised Gonadotropin -Releasing Hormone (GnRH) analogue treatment in the form of Leuprolide 7.5mg every four weekly but the patient could not afford it.
Discussion
Precocious puberty is defined as appearance of secondary sexual characters before the age of eight years in girls and nine years in boys [1]. CPP, also called true precocious puberty is caused by early activation of HPG axis [2]. It is idiopathic in more than 80% cases and is more common in girls [3]. Secondary causes of CPP include Central Nervous System (CNS) tumour (commonest tuber cinereum hamartoma), infection, congenital defect, radiation or injury [4]. PPP, also called pseudo-precocious puberty does not involve the HPG axis and is caused by release of sex steroid from adrenal, gonad or exogenous source or ectopic gonadotropin production from germ cell tumour. In CPP, testes are usually of pubertal or post-pubertal size and basal LH level is often raised with a pubertal response to GnRH stimulation. In PPP, testes size are usually pre-pubertal and basal LH level is low without any response to GnRH stimulation, germ cell tumour and familial testotoxicosis being two exceptions mimicking CPP [2]. GnRH stimulation test is done either by GnRH itself or by GnRH agonist. In our study, the test was done by 100 mcg subcutaneous tryptorelin injections and LH was measured 40 minutes and four hours after injection [5].
Our case is a six-year-old boy having precocious puberty apparently CPP with bilateral large testes (12 ml each), high serum testosterone level 599ng/dL, high triptorelin stimulated LH (29.38IU/mL in 40 minutes and 28.98IU/mL in four hours) but at the same time high 17-hydroxyprogesterone level (4050ng/dL) suggesting that originally it is a case of Congenital Adrenal Hyperplasia (CAH) with PPP converted to CPP due to prolonged sex steroid exposure. A case series containing three male child having CAH with CPP is comparable to our case which is more florid in presentation [6].
However, children with CAH might develop CPP with early maturation of HPG axis [7]. Our case presented initially like CPP with testicular growth and elevated gonadotropins but peculiar salt craving feature of the child indicated towards adrenal cause and subsequent work-up revealed undiagnosed CAH.
CAH is a group of autosomal recessive disorders resulting from the deficiency of one of the enzymes of adrenal steroidogenesis [Table/Fig-7], the most frequent (90%) being 21-hydroxylase. A 21-hydroxylase is a cytochrome P-450 enzyme which catalyzes the conversion of 17-hydroxyprogesterone to 11-deoxycortisol, a cortisol precursor, and progesterone to deoxycorticosterone, an aldosterone precursor. The resultant hypocortisolism increases corticotropin release from anterior pituitary which causes adrenal gland hyperplasia as well as overproduction of 17-hydroxyprogesterone and androgen. This is more pronounced when stimulated by gonadotropin releasing hormone analogue. Androgen excess is responsible for early virilization [8].
Adrenal steroidogenesis: SCC-side chain cleavage, 3βHSD2- 3β hydroxysteroid dehydrogenase, 17βHSD2- 17β hydroxysteroid dehydrogenase type 2.
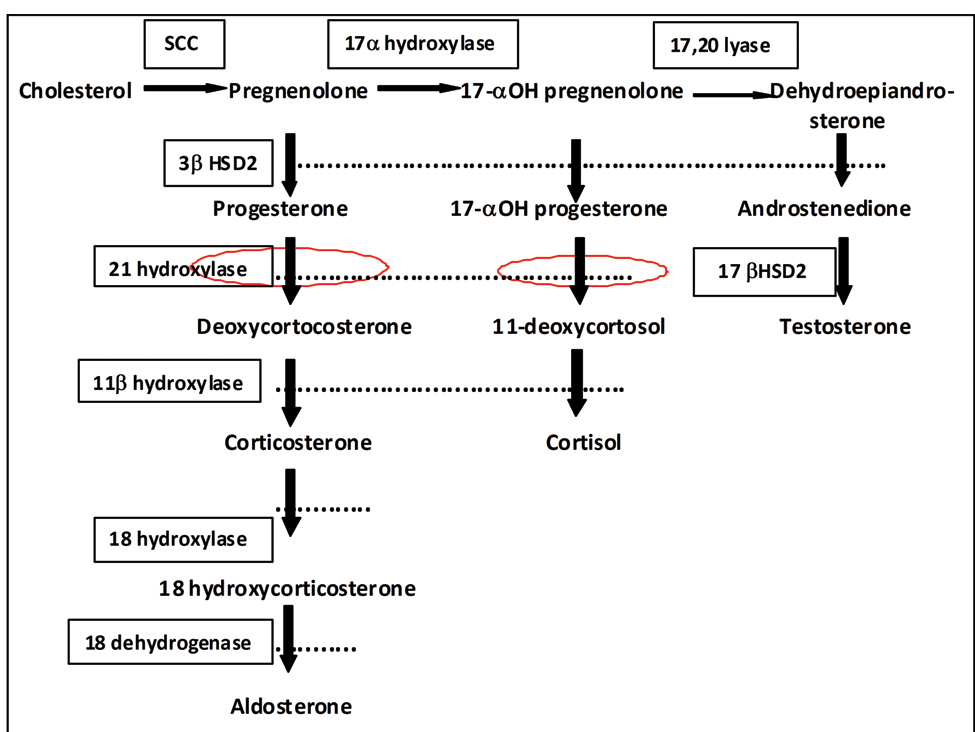
Classical form of CAH is subdivided into salt-losing and simple virilizing categories, depending on the presence or absence of mineralocorticoid deficiency respectively. Salt-losing form presents with neonatal adrenal crisis which is often fatal. Simple virilizing form manifests as ambiguous genitalia in female newborns whereas non-salt-losing form in males presents as precocious puberty in toddlers. In both sexes, increased androgen production leads to initial growth acceleration with reduced final height due to premature fusion of growth plates [9].
There are very few Indian studies showing prevalence of congenital adrenal hyperplasia. In Government Medical College, Chandigarh, out of 6813 neonates screened by estimation of 17α OH progesterone carried on dried blood spots using DELFIA (Dissociation Enhanced Lanthanide Fluorescent Immunoassay) method only one was found to be true positive for congenital adrenal hyperplasia. Exact data for late presentation leading to central precocious puberty was not available [10].
The goal of therapy in CAH with precocity is to replace the missing glucocorticoids thereby suppressing ACTH and normalizing adrenal androgens. Hydrocortisone in three divided doses is preferred because of its short half-life and lower likelihood of suppressing growth; usual starting dose being 10-15 mg/m2/day [11].
Our case presented like CPP, but further workup revealed increased DHEAS and 17-hydroxy progesterone and imaging evidence of adrenal hyperplasia. The child was treated with oral hydrocortisone which resulted in arrest of pubertal development and growth acceleration. The clinical, biochemical and radiological evidence as well as marked improvement on hydrocortisone therapy established the diagnosis of CAH presented with CPP.
Conclusion
It is evident from available data that CAH is the second most common neonatal disorder in India after congenital hypothyroidism [12]. The clinical and biochemical evidence of CPP with normal brain imaging in a male child should be considered for further evaluation to rule out CAH as timely diagnosis and proper intervention not only could arrest the puberty but can also save the life of the child from adrenal crisis during intercurrent illness.
[1]. Adashi EY, Rock JA, Rosenwaks Z, Reproductive Endocrinology, Surgery, and Technology 1996 Vol 1PhiladelphiaLippincott-Raven:989 [Google Scholar]
[2]. Berberoğlu M, Precocious Puberty and Normal Variant Puberty: Definition, etiology, diagnosis and current managementJournal of Clinical Research in Pediatric Endocrinology 2009 1(4):164-74. [Google Scholar]
[3]. Bridges NA, Christopher JA, Hindmarsh PC, Brook CG, Sexual precocity: sex incidence and aetiologyArch Dis Child 1994 70(2):116-18. [Google Scholar]
[4]. Fahmy JL, Kaminsky CK, Kaufman F, Nelson MD Jr, Parisi MT, The radiological approach to precocious pubertyBr J Radiol 2000 73(869):560-67. [Google Scholar]
[5]. Kumar M, Mukhopadhyay S, Dutta D, Challenges and controversies in diagnosis and management of gonadotropin dependent precocious puberty: An Indian perspectiveIndian Journal of Endocrinology and Metabolism 2015 19(2):228-35. [Google Scholar]
[6]. Veetil VM, Naseerali MC, Congenital adrenal hyperplasia-presenting as central precocious pubertyInternational Journal of Pediatric Endocrinology 2013 2013(Suppl 1):118 [Google Scholar]
[7]. Soliman AT, AILamki M, AISalmi I, Asfour M, Congenital adrenal hyperplasia complicated by central precocious puberty: linear growth during infancy and treatment with gonadotropin-releasing hormone analogMetabolism Clinical & Experimental 1997 46(5):513-17. [Google Scholar]
[8]. Speiser PW, White PC, Congenital adrenal hyperplasiaN Engl J Med 2003 349(8):776-88. [Google Scholar]
[9]. Carel JC, Lahlou N, Roger M, Chaussain JL, Precocious puberty and statural growthHum Reprod Update 2004 10(2):135-47. [Google Scholar]
[10]. Kaur G, Srivastav J, Jain S, Chawla D, Chavan BS, Atwal R, Preliminary report on neonatal screening for congenital hypothyroidism, congenital adrenal hyperplasia and glucose-6-phosphate dehydrogenase deficiency: a Chandigarh experienceIndian J Pediatr 2010 77(9):969-73. [Google Scholar]
[11]. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Congenital adrenal hyperplasia due to steroid 21- hydroxylase deficiency: an Endocrine Society clinical practice guidelineJ Clin Endocrinol Metab 2010 95(9):4133-60. [Google Scholar]
[12]. Kapoor S, Kabra M, Newborn screening in India: current perspectivesIndian Pediatrics 2010 47:219-24. [Google Scholar]