Clinical and Metabolic Profile of Glutaric Aciduria Type 1 from North India: Tertiary Centre Experience
Ankur Singh1, Rajniti Prasad2, Seema Kapoor3, Om Prakash Mishra4
1 Assistant Professor, Department of Paediatrics, IMS-BHU, Varanasi, Uttar Pradesh, India.
2 Director Professor, Department of Paediatrics, MAMC, New Delhi, India.
3 Professor, Department of Paediatrics, IMS-BHU, Varanasi, Uttar Pradesh, India.
4 Professor, Department of Paediatrics, IMS-BHU, Varanasi, Uttar Pradesh, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Ankur Singh, Assistant Professor, Department of Paediatrics, IMS-BHU, Varanasi-221005, Uttar Pradesh, India.
E-mail: pediaankur@gmail.com
Introduction
Glutaric aciduria type 1 is caused by deficiency of glutaryl-CoA dehydogenase leading to accumulation of glutarylcarnitine in blood and excretion of glutaric acid, 3-hyroxyglutaric acid and glutaconic acid in urine. It can be diagnosed through high risk screening in symptomatic cases.
Aim
To know the clinical, biochemical, neuroimaging and outcome profile of Glutaric aciduria type 1 patient diagnosed during testing by Tandem Mass Spectrometry (TMS) and Gas Chromatography and Mass Spectrometry (GCMS).
Materials and Methods
It was retrospective record analysis of patients diagnosed with Glutaric aciduria type 1. 2000 patients were screened for various indications like (developmental delay/ regression, unexplained seizures, encephalopathy, dystonia, chorea, large head, unexplained sibling death). Screening strategy involved estimation of lactate, ammonia, TMS and GCMS. Neuroimaging was done where it was required. This study was conducted over a period of three years (January 2014 to December 2016).
Results
Study group comprised of 10 males and 3 females. Median age (interquartile range) of presentation in study group was 11 months (10-22.5). Pretesting diagnosis was suspected as inborn error of metabolism in each case based on clinical presentation. Seizure and dystonia were important clinical presentation. Frontotemporal atrophy was important neuroimaging finding. Macrocephaly was present in two of thirteen cases. Glutarylcarnitine level was normal in 5 of 11 patients, suggesting poor sensitivity of TMS in diagnosed cases. There was wide variation in excretion of urinary metabolite from cases to cases, highlighting genetic heterogenousity.
Conclusion
Seizures and dystonia were important clinical presentations. Presence of bilateral frontotemporal atrophy in clinical testing was an important clue to diagnosis. Presence of macrocephaly (important sign of disease) was present in only two cases. There was only one death in follow up.
Glutaryl-CoA dehydrogenase gene, Glutaryl-CoA dehydrogenase/deficiency, Metabolic brain diseases
Introduction
Glutaric Aciduria (GA) is a neurometabolic disorder, characterized by seizure, dystonia macrocephaly and frontotemporal atrophy. It is inherited deficiency of glutaryl- CoA dehydrogenase due to mutation in glutaryl-CoA dehyrogenase gene, located on 19p13.2 [1]. The first patient was reported by Goodman SI et al., in 1975 [2]. This leads to defect in metabolism of lysine, hydroxylysine and tryptophan and accumulation of glutaric acid, 3-hydroxyglutaric acid and glutaconic acid in body fluids mainly affecting central nervous system. Estimated frequency of GA Type I is 1 in 30,000 to 100,000 newborns [3]. Majority of patients present with extrapyramidal symptoms predominantly dystonia, axial hypotonia, precipitated by intercurrent illlnesses like fever, diarrhoea, vaccination [4]. Since then, more than 500 patients have been reported worldwide. Acute neurological deterioration occurs between 6 and 18 months of age. Most patients have macrocephaly at birth or develop thereafter. Few patients can present with subdural hygroma or subdural haemorhage after trivial trauma [Table1/Fig-1].
MRI image (FLAIR axial section) showing generalised atrophy of brain parenchyma and subdural hygroma (arrows); b) MRI image (T2 weighted axial section) showing large hyperintense subdural hygromas (arrows).
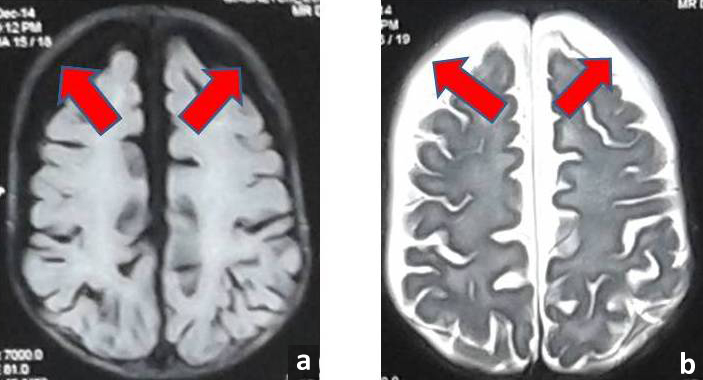
These patients usually present with fever, hypotonia, seizures after diarrhoeal episodes, viral illnesses, or immunization. Striatal injury precipitates the onset of dystonia in such patients. Episodes of acute decompensation have been reduced after 5 years of age. Most patients surviving in to adulthood require assistance and are wheel chair bound [5,6]. Present study was undertaken to address the extent of problem in our group of high risk newborn and to study clinical, biochemical, neuroimaging and outcome profile.
Materials and Methods
The patients were being referred to metabolic clinic after being suspected of having Inborn error of metabolism (IEM) as cause of seizure, global developmental delay/regression, dystonia, presence of consanguinosity, history of unexplained sibling death, history of recurrent abortion, unexplained encephalopathy. We screened 2000 patients for IEM over period of three years. These patients were symptomatic high risk cases. We found thirteen patients of Glutaric aciduria Type I who presented to genetic clinic of Lok Nayak hospital, New Delhi, India, between period from January 2014 to December 2016. Study was approved by Institute Ethical committee and informed consent was taken from parents. Clinical information, neuroimaging findings, metabolic workup were entered in predesigned performa. Metabolic work up included estimation of ammonia, lactate, Tandem mass spectroscopy and Gas chromatography mass spectroscopy. Estimation of lactate was done with following method: Blood was collected from the free vein and stored in an ice bath. The plasma was then separated by centrifugation within 20 minutes. We pipetted three cuvettes: one containing 5 μL of double distilled water and 500 μL enzyme reagent (Reagent Blank Solution), 5 μL of standard S1 with 500 μL of enzyme reagent (Standard S1), and finally third cuvette containing 5 μL of sample with 500 μL of enzyme reagent (Sample). We mixed and incubated for 10 minutes at +20°C to +25°C or 5 minutes at +37°C. We measured the absorbance at 550 nm of the sample (A sample) and standard (A standard) against the reagent blank within 5 minutes.
Ammonia was estimated with following method: blood was collected from the vein and stored in an ice bath. The plasma was then separated by centrifugation within 20 minutes. We pipetted three cuvettes: one containing 100 μL of double distilled water and 1500 μL enzyme reagent (Reagent Blank Solution); 100 μL of standard S1 with 1500 μL of enzyme reagent (Standard S1); and finally third cuvette containing 100 μL of sample with 1500 μL of enzyme reagent (Sample). We mixed and allowed to stand for 5 min. We read initial absorbance at 340 nm of Sample, Standard and Blank (A1). After this we added Enzyme reagent (R2: Glutamate Dehydrogenase) 10 μL to each of three cuvettes. Again, we mixed and allowed to stand for 5 min. We read final absorbance at 340 nm of Sample, Standard and Blank (A2). The corresponding decrease in absorbance at 340 nm was proportional to the plasma ammonia concentration. Tandem Mass Spectrometry (TMS) was done on dried blood spot. Urine samples were subjected to estimation with help of Gas Chromatography and Mass Spectrometry (GCMS), following a standard protocol [7]. Before testing samples from high risk babies, reference range for various urinary metabolites were established in 100 healthy North Indian children in our population.
Results
Demographic characteristics, clinical features, neuroimaging and metabolic data were tabulated in [Table/Fig-2]. Median age for onset of symptoms was seven months (IQR: 6.5-8). Most common clinical, neuroimaging and metabolic features were: History of Consanguinity (9/13), seizures (9/13), dystonia (8/13), global developmental delay (predominantly motor and speech) (13/13) frontotemporal atrophy (8/11), glutaric aciduria (13/13) (Normal range: 0-31.2 mmol/mol of creatnine). Frequency of other features were macrocephaly (2/13), elevated ammonia (0/13), elevated lactate (4/11), elevated glutarylcarnitine on TMS (7/12) (Normal Range: 0.00- 0.56 μmol/L). Characteristic neuroimaging finding of fronto-temporal atrophy and subdural hygroma is seen in [Table/Fig-3].
Clinical, metabolic, neuroimaging and outcome profile of thirteen North Indian patients.
| Patient | Age (months) of onset of symptoms | Age (months) at time of present-ation | History of consan-gulty | Sex | HC (cm) | Seizure | EPS (Dystonia) | Develop-mental Delay | MRI/CT front otemporal atrophy and basal ganglia hyper intensity on T2 and FLAIR | Ammonia (15-45 microgram / deciliter) | Lactate (0.5 –1.5 mmol/L) | TMS (C5 DC 0.00-0.56 μmol/L) | GCMS (Glutaric acid: 0-31.2 mmol/mol ofcreatinine) | Duration of follow up in months |
|---|
| 1 | 6 | 10 | + | m | 46.5 | + | – | + | + | n | abn | 1.47 | 6065.88 | 20 |
| 2 | 6 | 11 | + | m | 43.5 | + | + | + | n | n | nd | 0.85 | 19745.79 | 21 |
| 3 | 7 | 10 | + | f | 42 | + (West syndrome) | – | + | n | n | n | 0.09 | 1255.6 | – |
| 4 | 5 | 6 | + | m | 40 | + | – | + | nd | n | abn | and | 48884.93 | 22 |
| 5 | 7 | 21 | + | m | 45 | – | + | + | nd | n | n | 1.56 | 4427.43 | 12 |
| 6 | 8 | 14 | + | m | 50 (> + 2z score) | + | + | + | + | n | n | 0.45 | 9498.79 | 10 |
| 7 | 8 | 24 | + | m | 53 (> + 2z score) | – | – | + | + | n | n | 0.08 | 773.65 | 8 |
| 8 | 7 | 21 | + | f | 45.5 | + | + | + | _ | n | abn | 0.09 | 10326.32 | 7 |
| 9 | 12 | 156 | + | m | – | – | + | + | + | n | abn | 1.02 | 5689 | 6 |
| 10 | 7 | 11 | – | m | 45 | + | + | + | + | n | n | 0.75 | 21140.9 | 20 |
| 11 | 8 | 36 | – | m | 52 | + | + | + | +(n subdural) | n | n | 2.79 | 12331.8 | 6 |
| 12 | 7 | 9 | – | m | 42 | + | + | + | + | n | n | 2.36 | 7971.17 | 22 |
| 13 | 7 | 10 | – | f | 44.5 | – | – | + | + | n | n | 0.45 | 20,000 | 24 |
MRI image (T1 weighted axial section) of a case showing prominence of fronto-temporal atrophy with bilateral open sylvian fissures (Giving characteristic Bat wing appearance).
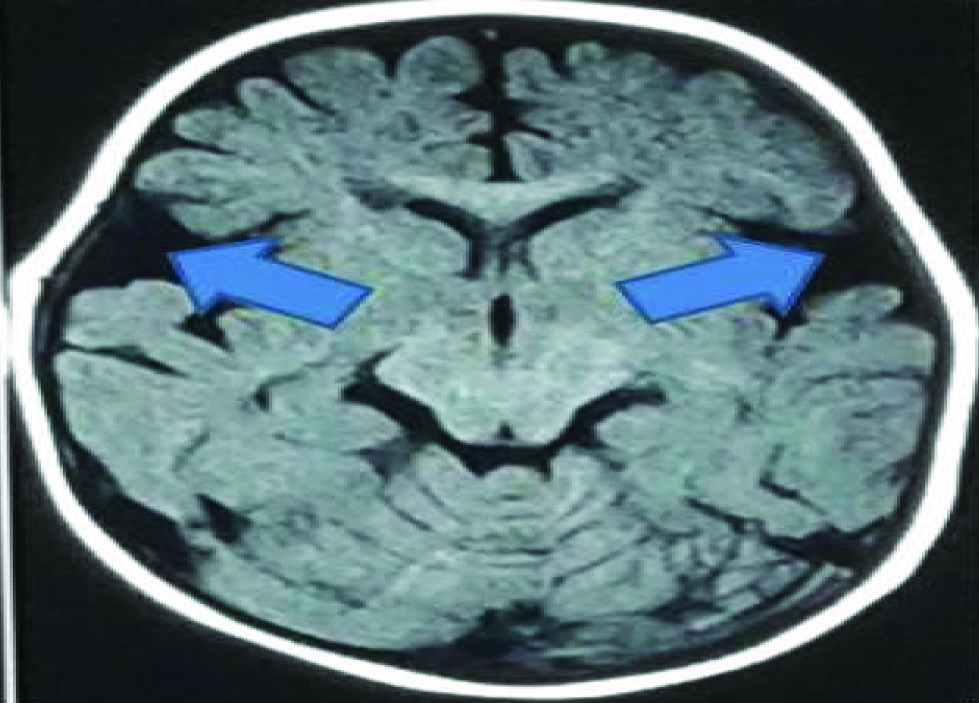
Median (IQR) duration of follow up in study group was: 16 (7.5-21.5) months. All patients were put on antiepileptic drugs, antidystonia drugs (Baclofen and benzodiaphines), lysine free/tryptophan protein substitutes (those who could afford), protein restriction, carnitine supplementation. Patients showed remarkable improvement in symptoms.
Discussion
Present study is retrospective data collection from patient’s genetic records. There was male preponderance of cases. There was one case who presented as west syndrome. TMS is a screening tool to diagnose most of inborn error of metabolism included in expanded list of Newborn screening. Glutaric aciduria type 1 is one of them. It is already included in Newborn screening programme of most of American and European countries to diagnose newborns in presymtomatic stage to provide maximum benefit by dietary manipulation [8]. GCMS is noninvasive diagnostic tool for many inborn errors of metabolism as accumulated toxic metabolites are excreted through urine and detected by their respective peaks in urinary chromatogram. Diagnosis was suspected in present cases based on clinical presentation, neuroimaging and confirmed by estimation of urinary metabolites glutaric acid. Glutarylcarnitinie (C5DC) was elevated in seven of twelve cases. It was normal in 5 cases but diagnosis was made by estimation of glutaric acid in urine. The explanation of normal C5DC in five patients was that their free carnitine was lower than normal thereby, leading to no elevation of C5DC in TMS. There was wide variation in excretion of urinary metabolite, confirming the presence of low excretors group in our ethnic population too. There is established genotype phenotype correlation for low excretors cohort [5]. We found no difference in clinical severity among low and high excretor patients, as previously found by Hedlund GL et al., [5]. Western countries are already utilising these newer technologies as part of their newborn screening programme. These technologies are present at very few centres in India. We are using this for diagnosing IEM in our high risk sick babies and thus, we are making diagnosis in symptomatic phase when maximum insult have already been done to many vital organs like brain. Recently Couce ML et al., and Viau K et al., have observed that early diagnosis have favourable outcome in GA Type I cases as against late diagnosis [9,10]. There are studies in various ethnic population citing early diagnosis, diagnosis by newborn screening and dietary compliance as the determinants of good outcome [11,12]. New born screening remains the optimal intervention to improve the timely outcome in late onset patients [13]. Two recent large Indian studies from North India (17 patients) and south India (11 patients) have highlighted clinical, biochemical and mutational profile [14,15]. Gupta N et al., highlighted following factors for determining the poor outcome of these patients: lack of new born screening programme, lack of awareness among clinicians, distance to appropriate medical facility, lack of home management and poor patient compliance due to illiteracy and socioeconomic status [14]. We compared our data with recently published Indian studies [14,15]. There is not much difference in age of presentation, clinical and neuroimaging profile of our patients as compared to two Indian studies. There was difference in elevated level of glutarylcarnitine. Our cohort has 5 patients who had C5DC in normal range as against these two Indian studies [14,15]. Macrocephaly was also present in only two cases as against 12/17 cases in North Indian patients [14]. Macrocephaly is not constant feature of disease. Similar finding of large head was reported by Morton DH et al., in only five of his 14 patients and 3 of 12 cases by Kyllerman M et al., [16,17]. The exact pathogenesis behind large head circumference is not known [16]. Normal neuroimaging finding in 2 patients can be explained by absence of any neurological crisis. Such finding has been previously reported by Bjugstad KB et al., in a cohort of 115 patients [18]. West syndrome patient was diagnosed based on triad of infantile spasm, disarrythmia (on EEG), and developmental delay. Neuroimaging finding in present case was normal which is not uncommon [19]. Pathological neuroimaging findings reported in west syndrome are cerebral malformations, cerebral atrophy, and delayed myelination [19]. This clearly highlights the variability in clinical presentation of this disorder. This change in phenotypic presentation may be attributed to changing genotype, epigenetic factors and nongenetic factors (infections, fasting, and fever).
Glutaric aciduria is caused by mutation in gene encoding glutaryl- CoA dehydogenase [5]. There are more than 200 mutations reported from different ethnic populations [20]. North Indian cohort studis by Gupta N et al., has concluded exon 11 and 8 as hot spot for Indian patients [14]. But no such hot spot was found in South Indian study by Radha Rama Devi A et al., [15]. As newborn screening is not mandatory in our country, onus of prenatal diagnosis lies on treating clinicians which is further based on correct clinical and molecular diagnosis in an affected case. The outcome of children diagnosed through newborn screening programme is very satisfying as against cases diagnosed clinically [21]. This emphasises the need of incorporating such treatable disorder in government sponsored programme in a phased manner to meet the challenges ahead.
Treatment of such disorder includes management of acute episodes with dietary supplement of carnitinie and lysine restricted diet. All cases were managed conservatively till they were diagnosed with GA type1 and put on special diet (low lysine) and carnitinine (50-100 mg/kg/day) [20,22], antidystonia drugs (Baclofen, Benzodiazepines) and antiepileptic drugs. Lysine restriction was done with help of special formula (lacking lysine and tryptophan). It was given to those who could afford. Help of dietician was taken in formulating diet plan for children. They were given multispecialty care in form of physiotherapy, speech therapy, dietary modification and genetic counselling. Children were followed up in special clinic. There was one death in follow up and rest 12 were alive. Most of them were symptom free (seizure and extrapyramidal features have subsided). and had started achieving developmental milestones.
Limitation
The study was done in high risk infants and children so we have missed the early asymptomatic cases. Since we did not have appropriate data on imaging features, We could not compare low excretors group with high excretors group with regard to neuroimaging parameter. We could not record dystonia scale data as it was not done. We could not do genetic studies on research basis due to which genotype-phenotype correlation was not possible. Genetic studies could have further shown the most prevalent mutation in cohort of patients.
Conclusion
Our study is the retrospective cohort of GA Type I patients from North India in which we have discussed the clinical and metabolic profile in detail. Global developmental delay, seizures and dystonia were important clinical presentation. Presence of bilateral frontotemporal atrophy provided important clue to clinicians to suspect GA Type I. Tools used for early diagnosis were: TMS and GCMS. Early diagnosis led to better management in all cases and helped families in genetic counselling.
[1]. Goodman SI, Stein DE, Schlesinger S, Christensen E, Schwartz M, Greenberg CR, Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (Type I): review and report of thirty novel mutationsHum Mutat 1998 12(3):141-44. [Google Scholar]
[2]. Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC, Glutaric aciduria; a “new” disorder of amino acid metabolismBiochem Med 1975 12(1):12-21. [Google Scholar]
[3]. Lindner M, Kölker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF, Neonatal screening for glutaryl-CoA dehydrogenase deficiencyJ Inherit Metab Dis 2004 27(6):851-9. [Google Scholar]
[4]. Hoffmann GF, Trefz FK, Barth PG, Böhles HJ, Biggemann B, Bremer HJ, Glutaryl-coenzyme A dehydrogenase deficiency: a distinct encephalopathyPediatrics 1991 Dec 88(6):1194-203. [Google Scholar]
[5]. Hedlund GL, Longo N, Pasquali M, Glutaric acidemia type 1Am J Med Genet C Semin Med Genet 2006 142C(2):86-94. [Google Scholar]
[6]. Strauss KA, Puffenberger EG, Robinson DL, Morton DH, Type I glutaric aciduria, part 1: natural history of 77 patientsAm J Med Genet C Semin Med Genet 2003 121C(1):38-52. [Google Scholar]
[7]. Mardens Y, Kumps A, Planchon C, Wurth C, Comparison of two extraction procedures for urinary organic acids prior to gas chromatography-mass spectrometryJ Chromatogr 1992 577(2):341-46. [Google Scholar]
[8]. Kölker S, Christensen E, Leonard JV, Greenberg CR, Burlina AB, Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria Type I)J Inherit Metab Dis 2007 30(1):5-22. [Google Scholar]
[9]. Couce ML, López-Suárez O, Bóveda MD, Castiñeiras DE, Cocho JA, García-Villoria J, Glutaric aciduria type I: outcome of patients with early- versus late-diagnosisEur J Paediatr Neurol 2013 17(4):383-89. [Google Scholar]
[10]. Viau K, Ernst SL, Vanzo RJ, Botto LD, Pasquali M, Longo N, Glutaric acidemia Type 1: outcomes before and after expanded newborn screeningMol Genet Metab 2012 106(4):430-38. [Google Scholar]
[11]. Tsai FC, Lee HJ, Wang AG, Hsieh SC, Lu YH, Lee MC, Experiences during newborn screening for glutaric aciduria type 1: Diagnosis, treatment, genotype, phenotype, and outcomesJ Chin Med Assoc 2017 80(4):253-61. [Google Scholar]
[12]. Lisyova J, Petrovic R, Jurickova K, Brennerova K, Urbanova D, Behulova D, GAI - distinct genotype and phenotype characteristics in reported Slovak patientsBratisl Lek Listy 2016 117(11):631-38. [Google Scholar]
[13]. Heringer J, Valayannopoulos V, Lund AM, Wijburg FA, Freisinger P, Barić I, Impact of age at onset and newborn screening on outcome in organic aciduriasJ Inherit Metab Dis 2016 39:341-53. [Google Scholar]
[14]. Gupta N, Singh PK, Kumar M, Shastri S, Gulati S, Kumar A, Glutaric acidemia type 1-clinico-molecular profile and novel mutations in gcdh gene in Indian patientsJIMD Rep 2015 21:45-55. [Google Scholar]
[15]. Radha Rama Devi A, Ramesh VA, Nagarajaram HA, Satish SP, Jayanthi U, Lingappa L, Spectrum of mutations in Glutaryl-CoA dehydrogenase gene in glutaric aciduria type I--Study from South IndiaBrain Dev 2016 38(1):54-60. [Google Scholar]
[16]. Morton DH, Bennett MJ, Seargeant LE, Nichter CA, Kelley RI, Glutaric aciduria type I: a common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, PennsylvaniaAm J Med Genet 1991 41(1):89-95. [Google Scholar]
[17]. Kyllerman M, Skjeldal OH, Lundberg M, Holme I, Jellum E, von Döbeln U, Dystonia and dyskinesia in glutaric aciduria type I: clinical heterogeneity and therapeutic considerationsMov Disord 1994 9(1):22-30. [Google Scholar]
[18]. Bjugstad KB, Goodman SI, Freed CR, Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type 1J Pediatr 2000 137(5):681-86. [Google Scholar]
[19]. Aydinli N, Calişkan M, Ozmen M, Tonguç E, Neuroradiologic aspects of West syndromePediatr Neurol 1998 19(3):211-16. [Google Scholar]
[20]. Kölker S, Christensen E, Leonard JV, Greenberg CR, Boneh A, Burlina AB, Diagnosis and management of glutaric aciduria type I--revised recommendationsJ Inherit Metab Dis 2011 34(3):677-94. [Google Scholar]
[21]. Lee CS, Chien YH, Peng SF, Cheng PW, Chang LM, Huang AC, Promising outcomes in glutaric aciduria type I patients detected by newborn screeningMetab Brain Dis 2013 28(1):61-67. [Google Scholar]
[22]. Boy N, Mühlhausen C, Maier EM, Heringer J, Assmann B, Burgard P, Proposed recommendations for diagnosing and managing individuals with glutaric aciduria Type I: second revisionJ Inherit Metab Dis 2017 40(1):75-101. [Google Scholar]