Supratentorial Haemangioblastoma without Von Hippel-Lindau Disease – A Rare Case Report with Review of Literature
Salman Tehran Shaikh1, Chandrashekhar Eknath Deopujari2
1 Neurosurgery Resident, Department of Neurosurgery, Bombay Hospital Institute of Medical Sciences, Mumbai, Maharashtra, India.
2 Head, Department of Neurosurgery, Bombay Hospital Institute of Medical Sciences, Mumbai, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Salman Tehran Shaikh, V-1, Row House II, Sector-6, Vashi, Navi Mumbai-400703, Maharashtra, India.
E-mail: shaikhsalman25@gmail.com
Haemangioblastomas are slow-growing, benign and vascular neoplasms of the central nervous system. They are usually infratentorial, occurring either sporadically in approximately 67% of cases or as a manifestation of Von Hippel-Lindau (VHL) disease in approximately 33% of cases. They were first described by Von Hippel in 1895. Haemangioblastoma in the supratentorial compartment is an infrequent occurrence. Only 58 cases of supratentorial haemangioblastoma without Von Hippel-Lindau disease have been reported from 1902 to 2015. This case discusses a left basifrontal supratentorial haemangioblastoma occurring in a young female who presented with headache and blurring of vision without manifestations of VHL disease. Its benign nature and prognosis merited surgical excision as the treatment of choice.
Haemangioblastoma, Supratentorial, Von hippel-lindau disease
Case Report
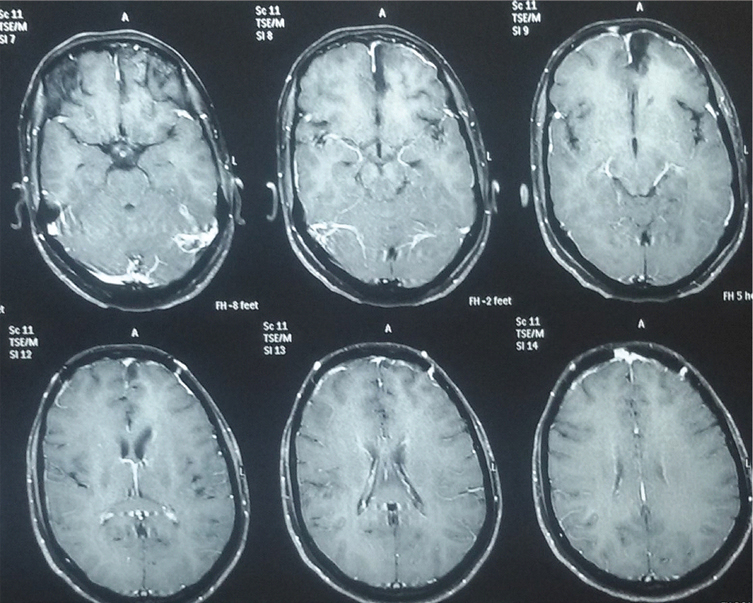
A 27-year-old female with history of hypothyroidism presented to the department of neurosurgery with complaints of frontal headache, blurring of vision with occasional diplopia for one month. There was no significant family history pertaining to her complaints. Neurological examination along-with visual acuity and fundus examination were normal. Magnetic Resonance Imaging (MRI) of brain (plain plus contrast) showed a solitary left basifrontal homogenously hyperintense lesion with hypointense rim on T2 Weighted image [Table/Fig-1], with perilesional white matter oedema on Fluid Attenuation Inversion Recovery Sequence (FLAIR) [Table/Fig-2]. Homogenous brilliant enhancement with a well-defined margin was seen with contrast administration [Table/Fig-3]. A differential diagnosis of glial neoplasm was suspected at this stage. A bifrontal craniotomy with gross total excision of the lesion was performed. Intra-operatively the lesion was firm, nodular, vascular and well circumscribed measuring approximately 2 cm x 2 cm in size. The lesion received branches from the fronto-polar artery. Histopathology was suggestive of a capillary haemangioblastoma with vessels lined by endothelium and interspersed stromal tumour cells [Table/Fig-4]. Immunohistochemistry examination was positive for Neuron Specific Enolase (NSE), Vimentin, CD 34, CD 31 with low MIB-1 index and was negative for epithelial membrane antigen and CD 10. This confirmed the vascular lining seen in haemangioblastomas and ruled out metastatic renal cell carcinoma. Postoperative recovery was uneventful with noticeable improvement in blurring of vision. Abdominal sonography and detailed visual examination was done to rule out concurrent Von Hippel-Lindau disease. She was discharged on anticonvulsants i.e., tablet levetiracetam 500 mg twice a day and was asked for follow up after three months. On Follow up after three months, she was asymptomatic and MRI showed complete excision of the lesion with no residue/recurrence [Table/Fig-5].
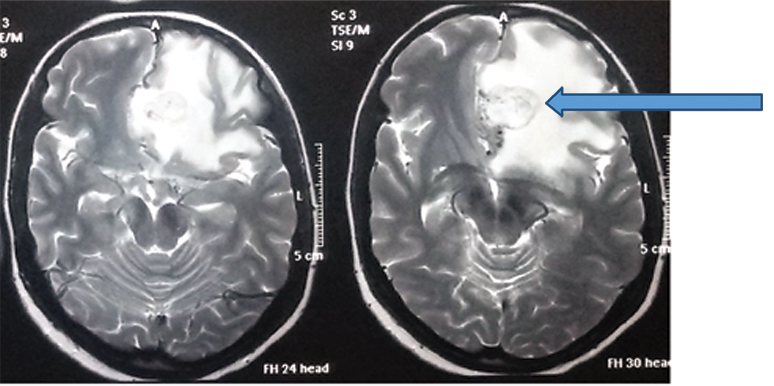
T2 Weighted axial MRI showing a solitary left basifrontal, homogenous, hyperintense lesion (arrow) with regular margins and a hypointense rim.
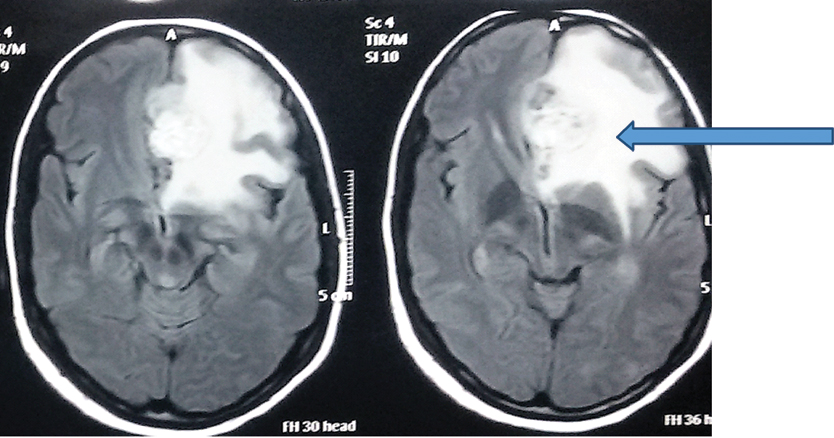
Perilesional white matter edema (arrow) seen on FLAIR MRI sequence.
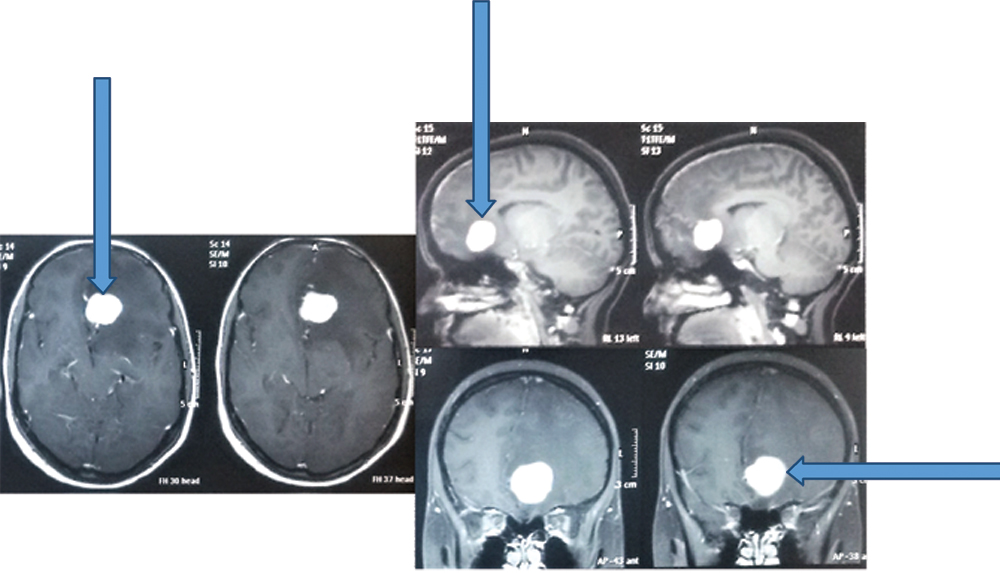
Contrast scans showing homogenous brilliant enhancement in the well- defined lesion (arrows).
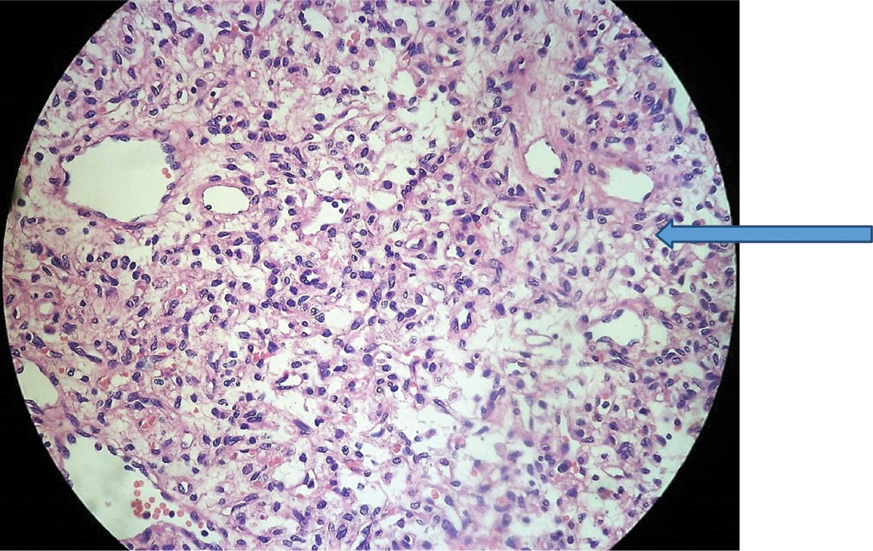
Histopathology image (Magnification: 40x) showing vessels lined by endothelium and interspersed stromal tumour cells (arrow).
Follow up MRI after three months showing complete excision.
Discussion
Haemangioblastomas account for 1.5% to 2.5 % of all intracranial neoplasms [1]. They are uncommon tumours of the central nervous system that were first described by Von Hippel in 1895 [2]. Bielschowsky first described supratentorial haemangioblastomas in the year 1902 [3]. Swedish pathologist Arvid Vilhelm Lindau first described cerebellar haemangioblastomas in 1926 and hence, they are frequently referred to as Lindau tumours [4]. In 1928, Cushing H and Bailey P introduced the term haemangioblastoma [5]. They are regarded as the most common primary adult intra-axial posterior fossa tumour [6]. The approximate incidence is 76% in the cerebellum around the fourth ventricle, 9% in cerebral hemispheres, 7% in spinal cord and 5% in brainstem [3]. Only 58 cases of supratentorial haemangioblastoma without VHL disease have been reported from 1902 to 2015 [3].
The latest WHO classification of tumours of the central nervous system in 2016 listed haemangioblastoma as a mesenchymal non-meningothelial tumour with behavior grading I i.e., of unspecified/borderline/uncertain behaviour [7]. On microscopy, they are composed of vascular components in the form of small capillaries with uniform endothelial cells and polygonal stromal vacuolated components which may be either cellular or more commonly of reticular subtype [8]. Genetic hallmark of this lesion is the loss of function of the VHL tumour suppressor protein on chromosome 3p. Immunohistochemistry markers for haemangioblastoma are inhibin-A, aquaporin1, NSE and Vimentin [9]. Haemangioblastomas appear either solid or cystic in consistency on gross appearance with a smaller mural nodule in 60 % of cases unlike astrocytomas [10]. Supratentorial haemangioblastomas tend to be associated with VHL disease in 60% of the cases and are more often solid in consistency [3]. The clinical features are not specific and are based upon the anatomical location of the lesion, peri-tumoural oedema, the rate of growth and its growth pattern. Accordingly, they may manifest as seizures, limb weakness, memory disturbances and/or slurring of speech. MRI with contrast is the imaging of choice as it has high resolution and sensitivity. The most common finding on imaging is the presence of a contrast enhancing mural nodule with an adjacent non-enhancing smooth cyst [11]. The possible differentials to be considered are angiomatous meningioma, glial neoplasm or metastasis from a clear cell variant of renal cell carcinoma [3,12]. The treatment modalities are complete surgical excision, gamma knife radio surgery, endovascular embolization and anti-angiogenic therapy. Surgery is curative, if complete excision is achieved as the lesion is benign and noninvasive. Resection of the tumour may be difficult in certain cases due to hypervascularity of the nidus and location of the tumour. To reduce the chances of severe bleeding in tumours, preoperative embolization of the solid component of the tumour has been attempted [13]. If the lesion is in the eloquent area then gamma knife radiosurgery or adjuvant radiotherapy may be considered. Sayer FT et al., reported the outcome at the end of three years of gamma knife radio surgery for 26 cases of intracranial haemangioblastomas in eloquent areas and observed that stereotactic radio surgery can achieve an acceptable level of preserved neurological function with an adequate control over the tumour growth in such patients [14]. Patients with multiple haemangioblastomas were poor candidates for long-term tumour control following radiosurgery. Antiangiogenic treatment employing Vascular Endothelial Growth Factor inhibitors is a treatment option in unresectable lesions. Schuch G et al., used antiangiogenic therapy with Semaxanib (tyrosine kinase receptor inhibitor) and found that soon after start of treatment, paresis resolved and preexisting hypaesthesia and dysaesthesia decreased [15]. Another report described by Aiello LP et al., showed a stable clinical remission in one patient with VHL disease and optic nerve haemangioblastomas treated with semaxanib over a period of 18 months [16]. The treatment of choice remains debatable with no widespread consensus due to its rarity of occurrence. It has been documented that supratentorial haemangioblastomas show a greater predisposition to grow during follow up (92.9%) as compared to their infratentorial or spinal cord counterparts (44%) [2]. MRI is advised for follow-up along with visual examination and abdominal imaging to watch for manifestations of VHL. In cases with concurrent VHL disease, lifelong follow up is recommended.
Conclusion
Haemangioblastoma in the supratentorial compartment is an uncommon occurrence. Paucity of available literature makes reporting of supratentorial haemangioblastoma cases helpful for better understanding of its clinical, pathological and radiological features. A high index of suspicion should be kept to rule out concurrent VHL disease in such cases.
[1]. Hussein MR, Central nervous system capillary haemangioblastoma: the pathologist’s viewpointInternational Journal of Experimental Pathology 2007 88(5):311-24. [Google Scholar]
[2]. Mills SA, Oh MC, Rutkowski MJ, Sughrue ME, Barani IJ, Parsa AT, Supratentorial hemangioblastoma: clinical features, prognosis, and predictive value of location for von Hippel–Lindau diseaseNeuro-Oncology 2012 14(8):1097-104. [Google Scholar]
[3]. Pandey S, Sharma V, Pandey D, Kumar V, Kumar M, Supratentorial haemangioblastoma without von Hippel–Lindau syndrome in an adult: A rare tumour with review of literatureAsian Journal of Neurosurgery 2016 11(1):8-14. [Google Scholar]
[4]. Lindau A, Studien uber Kleinhirnzysten. Bau, Pathogenese und Beziehung zur Angiomatosis retinaeActa Path et Microbiol Scand 1926 (Suppl 1):1-126. [Google Scholar]
[5]. Cushing H, Bailey P, Tumours arising from blood vessels of the brain: angiomatous malformations and haemangioblastomas 1928 Springfield, IIICharles C Thomas Publisher [Google Scholar]
[6]. Constans JP, Meder F, Maiuri F, Donzelli R, Spaziante R, de Divitiis E, Posterior fossa hemangioblastomasSurg Neurol 1986 25(3):269-75. [Google Scholar]
[7]. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, The 2016 World Health Organization Classification of Tumours of the Central Nervous System: a summaryActa Neuropathologica 2016 131(6):803-20. [Google Scholar]
[8]. Rubinstein LJ, Atlas of Tumour Pathology: Tumours of the Central Nervous System 1972 Washington, DCUS Government Printing Office:235 [Google Scholar]
[9]. Weinbreck N, Marie B, Bressenot A, Montagne K, Joud A, Baumann C, Immunohistochemical markers to distinguish between haemangioblastoma and metastatic clear-cell renal cell carcinoma in the brain: utility of aquaporin1 combined with cytokeratin AE1/AE3 immunostainingAm J Surg Pathol 2008 32(7):1051-59. [Google Scholar]
[10]. Ho VB, Smirniotopoulos JG, Murphy FM, Rushing EJ, Radiologic-pathologic correlation: haemangioblastomaAJNR Am J Neuroradiol 1992 13(5):1343-52. [Google Scholar]
[11]. Kim CS, Balk SK, Kim JD, Chung CP, Cho MY, Choi SS, MRI Findings of Intracranial HemangioblastomaJ Korean Radiol Soc 1995 33(5):705-11. [Google Scholar]
[12]. Sun Z, Yuan D, Sun Y, Yan P, Zuo H, Surgical resection of cerebellar hemangioblastoma with enhanced wall thickness: A report of two casesOncology Letters 2015 9(4):1597-99. [Google Scholar]
[13]. Liu AH, Peng TM, Wu Z, Xiao XR, Jiang CH, Wu ZX, Clinical effectiveness of preoperative embolization for cerebellar haemangioblastomaAsian Pac J Cancer Prev 2013 14(9):5179-83. [Google Scholar]
[14]. Sayer FT, Nguyen J, Starke RM, Yen CP, Sheehan JP, Gamma knife radiosurgery for intracranial haemangioblastomas-outcome at three yearsWorld Neurosurg 2011 75(1):99-105.discussion 45-8 [Google Scholar]
[15]. Schuch G, de Wit M, Höltje J, Laack E, Schilling G, Hossfeld DK, Case 2. Haemangioblastomas: diagnosis of Von Hippel-Lindau disease and antiangiogenic treatment with SU5416J Clin Oncol 2005 23(15):3624-26. [Google Scholar]
[16]. Aiello LP, George DJ, Cahill MT, Wong JS, Cavallerano J, Hannah AL, Rapid and durable recovery of visual function in a patient with Von hippel-lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416Ophthalmology 2002 109(9):1745-51. [Google Scholar]