Thalassaemia Trait with Gaucher Disease: A Diagnostic Dilemma
Jyoti Ramnath Kini1, Saraswathy Sreeram2, Anupama Hegde3, Sowmini Kamath4, Radha Ramachandra Pai5
1 Associate Professor, Department of Pathology, Kasturba Medical College, Manipal University, Mangaluru, Karnataka, India.
2 Assistant Professor, Department of Pathology, Kasturba Medical College, Manipal University, Mangaluru, Karnataka, India.
3 Associate Professor, Department of Biochemistry, Kasturba Medical College, Manipal University, Mangaluru, Karnataka, India.
4 Associate Professor, Department of Paediatrics, Kasturba Medical College, Manipal University, Mangaluru, Karnataka, India.
5 Professor, Department of Pathology, Kasturba Medical College, Manipal University, Mangaluru, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Saraswathy Sreeram, Assistant Professor, Department of Pathology, Kasturba Medical College, Lighthouse Hill Road, Hampankatta, Mangaluru-575001, Karnataka, India.
E-mail: swameeram@gmail.com
Gaucher Disease is an autosomal recessive disease caused by the accumulation of glucocerebrosidase due to deficiency in lysosomal glucocerebrosidase. Thalassaemia trait is asymptomatic and is usually an incidental diagnosis. Both thalassaemia and Gaucher disease can have similar haematologic manifestations and hence, their coexistence causes diagnostic dilemma. Our patient presented at one-and-a-half years with weakness, pallor, failure to thrive and massive hepatosplenomegaly. Clinical examination and history pointed to a lipid storage disease. Peripheral smear revealed microcytic hypochromic cells and nucleated red cells with haemolytic blood picture. Thalassaemia trait was indicated on haemoglobin variant analysis using High Performance Liquid Chromatography. Liver biopsy, bone marrow aspirate and therapeutic splenectomy revealed Gaucher-like cells. Type 1 Gaucher disease can be clinically asymptomatic as well as present with massive liver and spleen enlargement and involvement of bone marrow. Anaemia, splenomegaly and thrombocytopenia are the usual presentations at diagnosis, similar to the haemoglobinopathies. Gaucher-like cells with normal beta-glucocerebrosidase (pseudo-Gaucher cells) are seen in thalassaemia, leukaemia, mycobacterial infections and myeloma. Gaucher disease coexisting with thalassaemia trait is uncommon. We report the occurrence of thalassaemia trait and Gaucher disease in a child, which resulted in confusion regarding the haematological diagnosis. This report highlights the necessity of independent establishment of the diagnosis in every patient so that appropriate management decisions are taken.
Haemoglobinopathies, Hepatosplenomegaly, Storage disorders
Case Report
Our patient was born out of a non-consanguineous marriage and the boy presented at one-and-a-half-years of age with weakness, failure to thrive, delay in developmental milestones and abdominal distention. Examination revealed pallor and massive hepatosplenomegaly. A clinical diagnosis of lipid storage disease was made.
His routine blood investigations showed anaemia (Hb-7.4 g/dL) with low haematocrit of 32.3% and thrombocytopenia (90,000/cu.mm). Peripheral smear exhibited a microcytic hypochromic picture accompanied by occasional target cells and nucleated red blood cells with a mild degree of haemolysis. Haemoglobin variant analysis by High Performance Liquid Chromatography (HPLC) revealed 86.7% Hb A, 5.3% Hb A2 and 5.7% Hb F [Table/Fig-1a]. This pattern was suggestive of a Thalassaemia trait.
(a) Haemoglobin electrophoresis confirming Thalassaemia trait; (b) Gaucher cell in bone marrow aspirate – large cell with abundant “crumpled tissue paper” cytoplasm (Leishman stain, 100X oil immersion); (c) Liver biopsy showing sheets of Gaucher cells (Haematoxylin & Eosin stain, 40X); (d) Glucocerebroside staining positive on special stain in the liver biopsy (Periodic Acid Schiff stain, 40X); (e) Gross appearance of spleen in Gaucher disease – pale, homogenous, firm; (f) Scrape smear cytology of spleen – numerous singly scattered Gaucher cells (H & E stain, 20X); (g) Cytology of Gaucher cells in scrape smear of spleen – histiocytes with pale pink fibrillary cytoplasm (H&E stain, 40X); (h) Histopathology of spleen – splenic capsule with parenchyma almost entirely replaced by Gaucher cells (H&E stain, 10X); (i) Close-up view of histology of sheets of Gaucher cells in spleen – splenic sinusoids with RBCs seen (H&E stain, 40X).
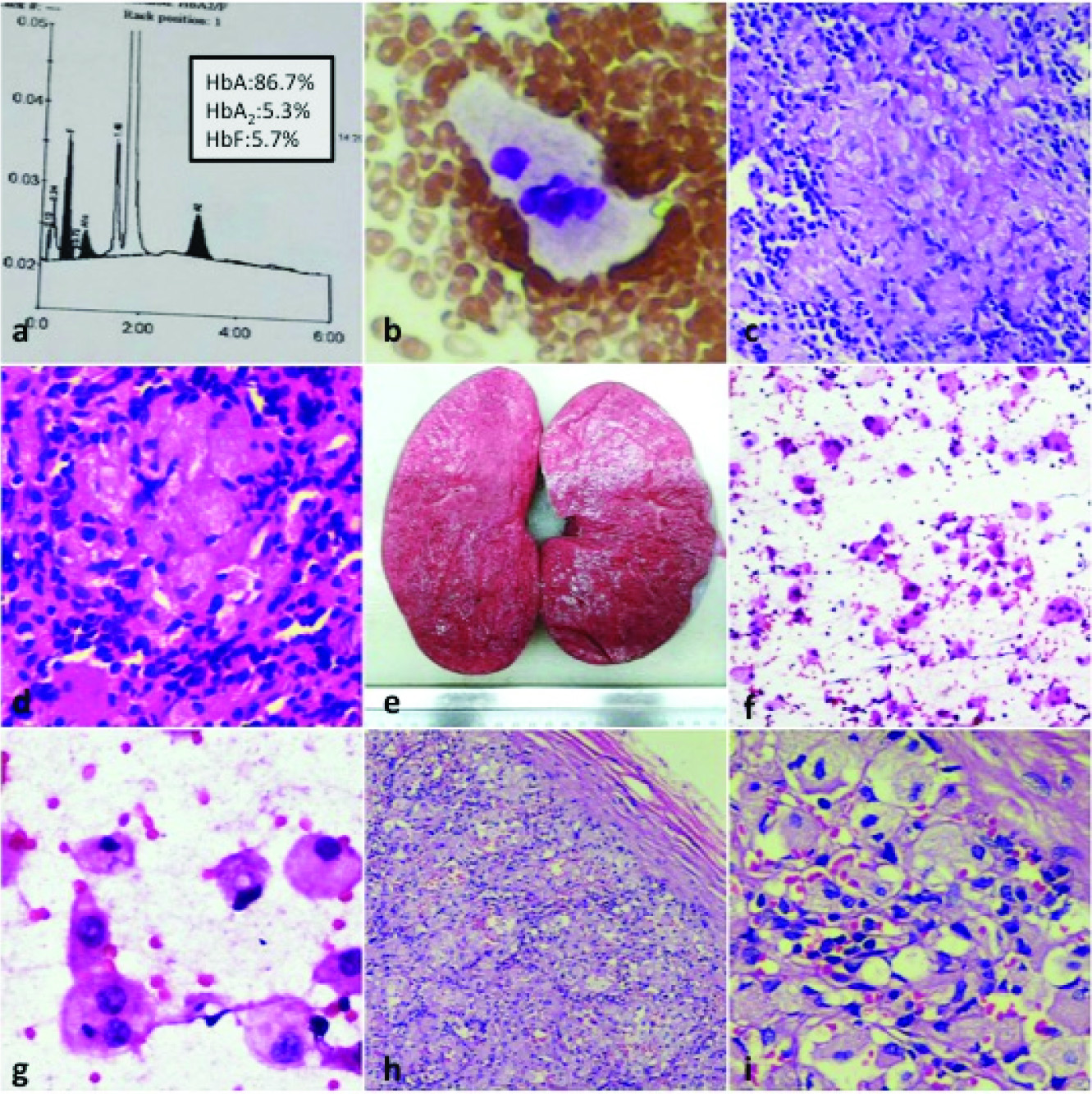
Bone marrow aspiration was done in view of failure to thrive and it revealed cells with fibrillary cytoplasm [Table/Fig-1b] and a possibility of Gaucher disease was suggested in the report. Liver biopsy was done for evaluation and it showed the same large cells with fibrillary cytoplasm [Table/Fig-1c]. These cells stained strongly positive for Periodic Acid Schiff (PAS) [Table/Fig-1d]. Therapeutic splenectomy was performed in the child because of the massive splenomegaly. Spleen weighed 740 grams and measured 17 x 10 x 9 cm. Cut section revealed a homogenous pink and firm surface [Table/Fig-1e]. Scrape smear cytology was done which showed numerous large cells with the same morphology as the cells in the bone marrow and liver biopsy [Table/Fig-1f,g]. Histopathological examination showed splenic parenchyma filled with cells having abundant eosinophilic granular cytoplasm and eccentric nuclei [Table/Fig-1h,i]. This supported the diagnosis of Gaucher disease. The collective findings revealed a case of coexistent Thalassaemia trait with Gaucher disease. The patient is on regular follow up since the splenectomy and he is doing well.
Discussion
The thalassaemias are a group of disorders characterized by decreased production of α- or β-globin chains of haemoglobin. Hypochromic, microcytic erythrocytes are seen due to a decreased production of haemoglobin in thalassaemia. The thalassaemias are extremely heterogeneous, both genetically and clinically. Thalassaemia trait is an asymptomatic disease and is usually an incidental diagnosis [1].
The overall prevalence of β-Thalassaemia Trait (BTT) in India is 2.78% with a range of 1.48-3.64% [2]. Thalassaemia trait is clinically innocuous, manifesting as RBC microcytosis with mild or no anaemia and occasional target cells. Coarse basophilic stippling is seen frequently. These patients do not need any medical intervention. The hallmark of thalassaemia trait is an increase in Hb A2 above 3.5% [1]. Our patient had Hb A2 level of 5.3%. It is significant to distinguish thalassaemia trait and provide appropriate genetic counselling to hopeful parents in order to prevent birth of a child with thalassaemia major and screen the remaining family members for presence of the disease.
Molecular analysis of the globin chain using Polymerase Chain Reaction (PCR) based assays and sequencing of PCR-amplified HBB gene are considered the most definitive for diagnosis of BTT [3]. Due to financial constraints, this test was not done in our subject.
Gaucher Disease is an autosomal recessive disease due to the accumulation of glucocerebrosidase in the cells of macrophagemonocyte system as a result of a deficiency in lysosomal glucocerebrosidase. This enzyme normally cleaves glucose residue and ceramide. A gene on chromosome 1q21 encodes this enzyme. A defect in the enzyme causes accumulation of glucocerebroside mainly in phagocytes, occasionally also in the central nervous system. It is the most common lysosomal storage disease and has varied clinical manifestations [4]. Three subtypes of Gaucher disease have been detected clinically [5].
Type I (chronic nonneuronopathic) is the most common form (99%). Spleen and skeletal system are the most commonly affected. Brain is not involved and glucocerebrosides accumulate in the phagocytes all over the body. Type II (acute neuronopathic) affects the infants and involves the brain. Type III is a transitional form between types II and I. The phagocytic cells with accumulations are known as Gaucher cells. They are found in the spleen, bone marrow, liver and lymphoid organs [6].
The spleen may be enlarged as much as 10 kg in Type I disease. As much as 70-100% cases show bone marrow involvement. The disease manifests only in adult life and clinical features are mainly related to marrow and splenic involvement. Hypersplenism leads to pancytopenia or thrombocytopenia. These findings are seen in haemoglobinopathies as well [5,6].
Gaucher disease can be diagnosed by measuring activity of glucocerebrosidase in white blood cells or in cultured skin fibroblasts [7]. The mainstay of treatment in Gaucher disease is replacement therapy with recombinant enzymes. This treatment is especially useful in Type I disease. Currently, the cost of this therapy is exorbitant [4-6].
Simultaneous occurrence of the two conditions causes a diagnostic dilemma due to some overlapping haematological features [8]. Our patient was diagnosed of this trait at a very young age and bone marrow aspiration, therapeutic splenectomy and liver biopsy showed Gaucher-like cells. Gaucher cells are large in size with cytoplasm showing a crumpled tissue paper appearance and cell nuclei are small and eccentric [1,5,6].
Pseudo-Gaucher cells are seen in Hodgkin’s lymphoma, acute lymphoblastic leukaemia, multiple myeloma, thalassaemia major and mycobacterial infections [9,10]. However, peripheral smear, bone marrow aspirate and biopsies of our patient failed to show any abnormal lymphoma or leukaemia cells. Pseudo-Gaucher is not associated with massive splenomegaly, as in our case. Presentation at a very young age is also a feature in favor of Gaucher disease. The pseudo-Gaucher cells stain negative for iron and ultra structurally lack cytoplasmic inclusions seen in Gaucher cells [9]. Perl stain for iron and Ziehl-Neelsen stain for mycobacteria were negative in our case, further tilting the balance in favour of Gaucher disease over pseudo-Gaucher cell-associated conditions.
The bone marrow, spleen and liver in this case showed presence of Gaucher cells. Our case report focuses on the simultaneous occurrence of thalassaemia trait with Gaucher disease, which is very unusual and can lead to a confusion regarding the correct diagnosis and further management.
Conclusion
Thalassaemia trait and Gaucher disease show a few similar clinical and laboratory features. In this report, we focus attention on contemporaneity of these entities. The association of haemoglobinopathies and storage disorders have been documented previously, however, coexistence as in our case has not been recorded so far in literature, to the best of our knowledge. Simultaneous occurrence of both these disorders should be kept in mind and appropriate investigations to prove the same must be done so that treatment can be planned accordingly.
[1]. Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS, Genetic disordersIn: Robbins and Cotran Pathologic Basis of Disease 2015 PhiladelphiaElsevier Saunders:153-4. [Google Scholar]
[2]. Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, Prevalence of β-thalassaemia and other haemoglobinopathies in six cities in India: a multicentre studyJ Community Genet 2013 4(1):33-42. [Google Scholar]
[3]. Brancaleoni V, Di Pierro E, Motta I, Cappellini MD, Laboratory diagnosis of thalassaemiaInt J Lab Haematol 2016 38(1):32-40. [Google Scholar]
[4]. Grabowski GA, Lysosomal storage diseases. In: Braunwald E, Fauci AS (eds)Harrison’s Principles of Internal Medicine 2001 15th EdNew York, NYMcGraw-Hill:2276-81. [Google Scholar]
[5]. Hruska KS, LaMarca ME, Scott CR, Sidransky E, Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA)Hum Mutat 2008 29:567-83. [Google Scholar]
[6]. Binesh F, Yousefi A, Ordooei M, Bagherinasab M, Gaucher’s Disease, an unusual cause of massive splenomegaly, a case reportIran J Ped Haematol Oncol 2013 3(4):173-5.Epub 2013 Oct 22 [Google Scholar]
[7]. Urban DJ, Zheng W, Goker-Alpan O, Jadhav A, Lamarca ME, Inglese J, Optimization and validation of two miniaturized glucocerebrosidase enzyme assays for high throughput screeningComb Chem High Throughput Screen 2008 11:817-24. [Google Scholar]
[8]. Miri-Moghaddam E, Velayati A, Naderi M, Tayebi N, Sidransky E, Coinheritance of Gaucher disease and α-thalassaemia resulting in confusion between two inherited haematologic diseasesBlood Cells Mol Dis 2011 46(1):88-91. [Google Scholar]
[9]. Chatterjee T, Dewan K, Ganguli P, Das S, Sharma A, Sahni AK, A rare case of haemoglobin E haemoglobinopathy with gaucher’s diseaseIndian J Haematol Blood Transfus 2013 29(2):110-2.Epub 2012 Apr 18 [Google Scholar]
[10]. Dunn P, Kuo MC, Sun JCF, Pseudo-Gaucher cells in mycobacterial infection: a report of two casesClin Pathol 2005 58(10):1113-4. [Google Scholar]