Audit of Organic Acidurias from a Single Centre: Clinical and Metabolic Profile at Presentation with Long Term Outcome
Seema Pavaman Sindgikar1, Krithika Damodar Shenoy2, Nutan Kamath3, Rathika Shenoy4
1 Associate Professor, Department of Paediatrics, K S Hegde Medical Academy, NITTE University, Mangalore, Karnataka, India.
2 Intern, Department of Paediatrics, Kasturba Medical College, Manipal University, Mangalore, Karnataka, India.
3 Professor, Department of Paediatrics, Kasturba Medical College, Manipal University, Mangalore, Karnataka, India.
4 Professor, Department of Paediatrics, K S Hegde Medical Academy, NITTE University, Mangalore, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rathika Shenoy, Professor, Department of Paediatrics, K S Hegde Medical Academy, Deralakatte, Mangalore-575018, Karnataka, India.
E-mail: rathika.shenoy@nitte.edu.in
Introduction
Organic Acidurias (OA) accounts between 10% and 40% of confirmed Inborn Errors of Metabolism (IEM) in India. With prompt recognition and management, better survival but adverse neurodevelopmental outcome is reported.
Aim
To study the clinical and metabolic presentation, management with immediate and long term outcome of symptomatic children with confirmed OA.
Materials and Methods
Hospital based study of symptomatic children diagnosed to have OA between 2003 and 2009 and the survivors followed up over next five years. Diagnosis was based on clinical and metabolic presentation and confirmed by spectrometry analyses of urine and blood. Management, immediate outcome, compliance to treatment and recurrence of crises were documented. Neurodevelopmental outcome was assessed in follow up. Mean with Standard Error (Mean ± SE) and frequencies with percentages were calculated.
Results
Of 72 cases suspected to have IEM, 38 (52.8%) were confirmed of (IEM), and out of which 15 (39.5%) had OA. Methyl malonic acidemia, multiple carboxylase deficiency and Propionic Acidemia (PA) constituted the largest proportion. Neurodevelopmental issues (73.3%) and metabolic crisis (53.3%) were common presenting features. Mean ± SE of ammonia was 639.0±424.1 μg/dl and lactate was 33.6±4.9 mg/dl. Mean pH, bicarbonate, and anion gap was 7.27±0.07, 14.1±2.3 and 17.9±2.3 respectively. Management was protocol based. Death was reported in two cases of PA; other morbidities were seen in five. Recurrent crisis (46.7%) complicated the follow up in survivors. Spasticity, extrapyramidal movement disorder, intellectual subnormality, autism spectrum, attention deficit hyperactivity disorder and sensory neural deafness were seen amongst survivors, in spite of compliance to therapy.
Conclusion
OA is part of differential diagnosis in sick children and treatment needs to be prompt and specific. Prognosis is guarded even with long term cofactor supplementation in the symptomatic.
Hyperammonemia, Metabolic acidosis, Neuropsychiatric issues, Sensory neural deafness
Introduction
IEM constitute an important and expanding group of monogenic disorders resulting from deficiency of a critical enzyme in the intermediary pathways of carbohydrate, protein and lipid metabolism or deficiency of a cofactor which is responsible for activation of an apoenzyme [1]. They offer formidable challenges to clinicians because of the myriad enzyme defects that exist, complexity of metabolic pathways, diverse clinical and metabolic manifestations, ability to mimic common pediatric illnesses, morbidity and implications in future pregnancies. In India, they occur more frequently because of high birth rate and consanguinity [2,3]. Compared to a decade ago, awareness and availability of specific investigations allow these disorders to be identified at much higher frequency as evidenced by several studies [3-6]. Although it has become imperative to move from an ’at risk’ or ’symptomatic sick child’ approach to a ’prospective’ or ’pre-symptomatic’ one, the services are availed by those affordable in the private sector. [7].
OA or organic acid disorders are a subgroup of IEM characterized by excretion of non amino organic acids in urine due to their accumulation in body fluids. Classic presentation of these disorders is with severe metabolic acidosis. Majority of OA are caused by defective catabolism of the three branched chain amino acids namely valine, leucine and isoleucine. OA accounts between 10% to 40% of confirmed IEM in India [8]. They are diagnosed by characteristic organic acid excretion pattern in urine Gas Chromatography Mass Spectrometry (GCMS). Disorders include Maple Syrup Urine Disease (MSUD), Isovaleric Acidemia (IVA), Propionic Acidemia (PA), Multiple Carboxylase Deficiency (MCD), Methyl Malonic Acidemia (MMA), beta Keto Thiolase (βKT) deficiency, Glutaric Aciduria type 1 (GA1) and others. MCD may result from biotinidase or holocarboxylase synthetase deficiencies with similar urine organic acid metabolites.
Prevention of death or permanent neurologic sequelae in patients with these disorders is dependent on early diagnosis and institution of appropriate therapy [9]. Most of these disorders are amenable to combination of dietary protein restriction, cofactor therapy with megavitamins and removal of toxic metabolites. During critical situation, suspicion as well as emergency treatment including metabolic stabilization will reduce mortality and morbidity. We present our data on the clinical and metabolic presentation, management, mortality and morbidity of symptomatic children confirmed to have OA by urine and blood spectrometry analyses and their five year follow up.
Materials and Methods
This is a hospital based study of children diagnosed with OA between 2003 and 2009 before Newborn Screening (NBS) was widely available. The survivors were followed up over the next five years or more. The study was cleared by the Institutional Ethics Committee. Clinical suspicion of an IEM was based on suspect family history, age, presentation, presence of a combination of failure to thrive, developmental delay or regression, recurrent vomiting, abnormal body fluid odours, encephalopathy and organomegaly. The metabolic work up included urine ketones, blood glucose, anion gap, arterial blood gas analysis, blood ammonia and lactate. Diagnosis was confirmed by urine GCMS for organic acids supplemented by blood tandem Mass Spectrometry (MS/MS). Management, immediate outcome, compliance to treatment and recurrence of crises were documented.
The survivors were prospectively followed up over next five years or more for growth and development, recurrence of symptoms, neurologic sequelae if any and neuroimaging. Failure To Thrive (FTT) was defined as weight, height and weight for age less than 5th centile in WHO growth standards. Developmental delay was defined as development quotient less than 70 and when possible intelligence quotient was assessed by Binet Kamat scale by a clinical psychologist. All children underwent vision and hearing assessment and where indicated brain imaging by Magnetic Resonance (MR).
Statistical Analysis
Statistical analyses included median or Mean±SE for numerical data and frequencies (n) with percentages (%) for categorical data.
Results
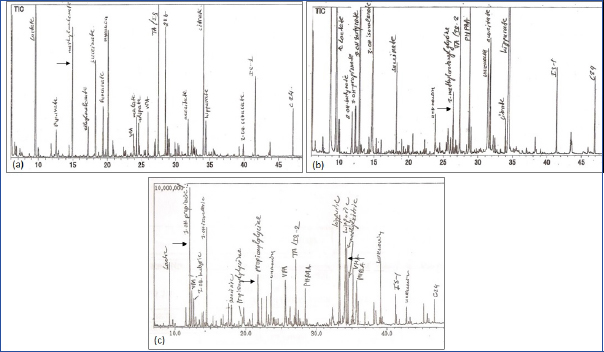
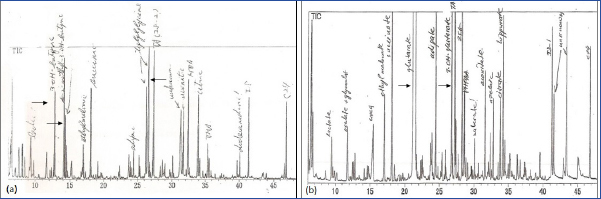
A diagnosis of IEM was considered in 72 children on the basis of clinical presentation and metabolic profile. Spectrometry analyses of urine and blood confirmed the same in 38 (52.8%) of which 15 (39.5%) had an OA. Except one child with MMA who was diagnosed at 14 months, all were diagnosed by infancy. The Mean±SE and median ages at diagnosis were 5.6±1.16 and 4 months respectively. Male preponderance was found with a ratio of 2:1. The most common OAs were MMA in 6 (40%) and MCD in 5 (33.3%). Others included two (13.3%) children of PA and one each of GA1 and βKT deficiency. [Table/Fig-1a-c,2a,b] depict the characteristic urine organic acid excretion pattern seen in these children. Consanguinity was seen in one and sibling affected/death in five (33.3%).
Urine gas chromatograph mass spectrometry showing characteristic organic acid excretion pattern: a): Methyl malonic acid in methylmalonic academia; b): 3-methyl crotonyl glycine in multiple carboxylase deficiency; c): 3-hydroxy propionic acid, propionyl glycine and methyl citrate in propionic acidemia. Absence of 3-hydroxy propionic acid and methyl citrate in a) is indicative of presentation without ketoacidosis.
Urine gas chromatograph mass spectrometry showing characteristic organic acid excretion pattern: a) 3-OH butyrate, 2-methyl 3-OH butyrate and tiglylglycine in beta ketothiolase deficiency; b) glutaric acid and 3-OH glutaric acid in glutaric aciduria type 1.
Developmental delay was the most common presentation seen in 73.3%. Developmental delay, seizures, alopecia and skin rash were diagnostic of MCD. Three children, one each of developmental delay with microcephaly, failure to thrive with seizures with no ketoacidosis and developmental delay with macrocephaly were confirmed to be MMA, PA and GA1 respectively. More than half (8,53.3%) presented with a metabolic crisis and symptoms at presentation included encephalopathy, seizures and rapid breathing due to metabolic acidosis. One each of MCD and MMA were confirmed by presymptomatic screening in view of sibling history. Two other children presenting as persistent vomiting were evaluated for gastro-oesophageal reflux disease and malrotation in surgical department before a diagnosis of MMA was made. The clinical presentation and metabolic profile at presentation are shown in [Table/Fig-3].
Clinical and metabolic profile of children (n=15) with confirmed organic acidemia at presentation.
| Clinical | Number(%) | Metabolic | Number(%) |
|---|
| Family history | 5 (33.3) | Elevated ammonia | 11 (73.3) |
| Recent weaning | 4 (26.7) | Elevated lactate | 9 (60.0) |
| Developmental delay | 11 (73.3) | Metabolic acidosis | 6 (40.0) |
| Encephalopathy | 8 (53.3) | Wide Anion gap | 8 (53.3) |
| Respiratory distress | 7 (46.7) | Respiratory alkalosis | 3 (20.0) |
| Failure to thrive | 5 (33.3) | Ketonuria | 6 (40.0) |
| Vomiting | 4 (26.7) | Hypoglycaemia | 3 (20.0) |
| Diarrhea | 3 (20.0) | Hyperglycaemia | 1 (6.7) |
| Hepatomegaly | 8 (53.3) | Hyponatraemia | 3 (20.0) |
| Skin lesions, alopecia | 4 (26.7) | | |
| Microcephaly | 1(6.7) | | |
| Macrocephaly | 1(6.7) | | |
Biochemically, wide anion gap metabolic acidosis, ketosis, lactic acidosis and hyperammonemia prompted the suspicion for diagnosis. The highest ammonia of 5706 μg/dl without ketoacidosis was seen in a child with PA [10]. The Mean ±SE ammonia level observed was 639.0±424.1 μg/dl (median 196 μg/dl) and lactate was 33.6±4.9 mg/dl. Mean pH was 7.27±0.07, arterial bicarbonate 14.1±2.3, base deficit 14.9±3.4 and anion gap 17.9±2.3. Neuroimaging (NI) was diagnostic in GA1 with bilateral widening of sylvian fissure and cerebrospinal fluid space anterior to temporal lobe (bat-wing appearance).
Empiric management in crisis included dehydration correction, megavitamins, carnitine, sodium benzoate when indicated, intravenous sodium bicarbonate based on base deficit in blood gases, broad spectrum antibiotics. Specific management after GCMS and MS/MS reports were available included protein restriction, cofactor supplementation like biotin for MCD and PA; vitamin B12 for MMA and riboflavin for GA1. The course was complicated by multiple crisis in 7 (46.7%) precipitated by infection. Poor compliance was noted only in one. The overall mortality observed was both of PA. Adverse outcome was seen in one of six MMA, three of five MCD and βKT deficiency. [Table/Fig-4] gives the adverse neurodevelopmental outcome in survivors of various categories of OA. Spasticity, extrapyramidal movement disorder, intellectual subnormality, autism spectrum, Attention Deficit Hyperactivity Disorder (ADHD) and sensory neural deafness were seen amongst survivors, in spite of compliance to therapy.
Summary of children with confirmed organic acidemia and adverse neurodevelopment outcome.
| Disorder | Age at diagnosis (months) | Presentation and Course | Outcome |
|---|
| Methyl malonic acidemia (1 of 6) | 4 | Presented as developmental delay, seizures and microcephaly No metabolic crisis Unresponsive to B12 | Spastic quadriplegia with severe microcephaly (Head circumference at 5 years:40 cm), severe psycho motor retardation, seizures, severe sensory neural hearingloss, failure to thrive. MR imaging: Gross cerebral atrophy with decreased whitematter and profound T2 hyperintensity suggestive of hypomyelination, coronalsuture craniosynostosis |
| Multiple carboxylase deficiency (3 of 5) | 2 | Metabolic acidosis, alopecia, intertrigo and seizures; no crisis on biotin | Spastic diplegia with attention deficit hyperactivity disorder, moderate intellectualdisability; off seizure medication. MR imaging: cerebral atrophy |
| 4 | Alopecia, intertrigo and seizures No crisis | Catch up in motor milestones, no tone abnormalities, mild intellectual disability, bilateral severe to profound sensory neural hearing loss with no languagedevelopment; off seizure medication |
| 8 | Metabolic acidosis in addition to alopecia, intertrigo and seizures; Poor compliance to therapy with recurrent crisis and seizures | Catch up in motor milestones, no tone abnormalities; complex partial seizureswith autism like and attention deficit hyperactivity disorder, moderate intellectualdisability, bilateral moderate sensorineural hearing loss; complex partial seizureson therapy.MR imaging: Normal |
| Beta keto thiolase deficiency (1) | 6 | Metabolic acidosis, hyperglycaemiaand encephalopathy; recurrent crisis | Extra pyramidal movement disorder, moderate intellectual disability; off seizuremedications. MR imaging: Normal |
Discussion
In our study, OA contributed to 20.8% of all children investigated and 39.5% of confirmed IEM. Among high risk children screened for IEM, Nagaraja D et al., and Narayanan MP et al., from Southern India have reported OA in 41.6 and 10.7% respectively [6,11]. Prevalence data of IEM in general and OA in particular from India is limited due to difficulties in establishing the diagnosis in an acutely sick child. Najafi R et al., have documented a high consanguinity rate of 84.7% in Mid-West [12]. In our study, though we did not have significant positive consanguinity, familial pattern in two suggested recessive inheritance pattern. High occurrence of MMA over other OA has been reported by other authors. Narayanan MP et al., and Najafi R et al., in their study report that MMA contributes to 34% and 33% of OA respectively [11,12]. Some of the unusual presentations in our children and reported as well, included PA masquerading as urea cycle disorder and βKT deficiency as diabetic ketoacidosis [10,13-17]. NI is highly useful in the diagnosis of GA1, as these children can have a subacute course and may present only with developmental delay and macrocephaly [18,19].
Neuropsychiatric manifestations occur in IEM either at presentation or as sequelae. It is thought to be due to disruption of normal late neurodevelopment or monoaminergic neurotransmitter systems [20]. A disturbance in mitochondrial function which is the final common pathway of all intermediary metabolisms may also be contributory. Autism spectrum disorder has been described in OA specifically PA, methylcrotonyl-CoA and biotinidase deficiencies [21-24]. The relation between ADHD and phenylketonuria is well documented [25]. ADHD is also associated with urea cycle disorders, maple syrup urine disease and homocystinuria. The autism and ADHD seen in our children with MCD may be part of moderate intellectual disability. NI was normal in this group.
Sensorineural hearing loss and optic atrophy are reported in untreated children with biotinidase deficiency [26-28]. It is proposed that accumulation of biocytin or small biotiny peptides may mediate neuronal injury in these children. Weber P et al., reported that vision and hearing abnormalities were none in infants diagnosed by NBS compared to children where treatment was initiated after onset of symptoms [26]. Wolf B et al., reports that more than three-fourths of symptomatic children have sensorineural hearing loss which does not seem to resolve or improve with biotin treatment [29]. Of the seven children with symptomatic biotinidase deficiency, Venkataraman V et al., report neurological deficit in one and deafness in none [30]. The variable prognosis in our series could be best explained by age and developmental period at diagnosis, severity of enzyme defect as well as phenotypic variations, recurrent metabolic decompensations, response and compliance to cofactor therapy.
Conclusion
Until NBS is a reality, OA should be part of the differential diagnosis in any sick neonate or infant with a negative sepsis screen, persistent metabolic acidosis and ketosis as well as in any infant with unexplained neurological deterioration even in the absence of ketoacidosis. Prognosis is guarded even with prompt institution of emergency treatment and long term cofactor supplementation.
[1]. Waber L, Inborn errors of metabolismPediatr Ann 1990 19(2):105-09.112-13, 117-18 [Google Scholar]
[2]. Choudhuri T, Sengupta S, Inborn error of metabolism - An Indian perspectiveInt J Hum Gen 2006 6(1):89-91. [Google Scholar]
[3]. Rao AN, Kavitha J, Koch M, Suresh Kumar V, Inborn errors of metabolism:review and data from a tertiary care centerInd J Clin Biochem 2009 24(3):215-22. [Google Scholar]
[4]. Devi ARR, Naushad SM, Newborn screening in IndiaIndian J Pediatr 2004 71(2):157-60. [Google Scholar]
[5]. Swarna M, Jyothy A, Rani PU, Reddy PP, Amino acid disorders in mental retardation:A two-decade study from Andhra PradeshBiochem Genet 2004 42(3–4):85-98. [Google Scholar]
[6]. Nagaraja D, Mamatha SN, De T, Christopher R, Screening for inborn errors of metabolism using automated electrospray tandem mass spectrometry:Study in high-risk Indian populationClin Biochem 2010 43(6):581-88. [Google Scholar]
[7]. Kapoor S, Kabra M, Newborn screening in India:Current perspectivesIndian Pediatr 2010 47(3):219-24. [Google Scholar]
[8]. Vaidyanathan K, Narayanan MP, Vasudevan DM, Organic acidurias:an updated reviewInd J Clin Biochem 2011 26(4):319-25. [Google Scholar]
[9]. Ozand PT, Gascon GG, Organic acidurias:a review. Part IJ Chil Neurol 1991 6:196-219. [Google Scholar]
[10]. Sindgikar SP, Rao S, Shenoy RD, Kamath N, Biochemical basis of heterogeneity in acute presentations of propionic acidemiaInd J Clin Biochem 2013 28(1):95-97. [Google Scholar]
[11]. Narayanan MP, Kannan V, Vinayan KP, Vasudevan DM, Diagnosis of major organic acidurias in children:Two years experience at a tertiary care centerInd J Clin Biochem 2011 26(4):347-53. [Google Scholar]
[12]. Najafi R, Hashemipour M, Mostofizadeh N, Ghazavi MR, Nasiri J, Shahsanai A, Demographic and clinical findings in pediatric patients affected by organic acidemiaIran J Child Neurol 2016 10(2):74-81. [Google Scholar]
[13]. Hori D, Hasegawa Y, Kimura M, Yang Y, Verma IC, Yamaguchi S, Clinical onset and prognosis of Asian children with organic acidemias, as detected by analysis of urinary organic acids using GC/MS, instead of mass screeningBrain Dev 2005 27(1):39-45. [Google Scholar]
[14]. Muranjan MN, Kondurkar P, Clinical features of organic acidemias:Experience at a tertiary care center in MumbaiIndian Pediatr 2001 38:518-24. [Google Scholar]
[15]. Akman I, Imamoğlu S, Demirkol M, Alpay H, Ozek E, Neonatal onset propionic acidemia without acidosis:a case reportTurk J Pediatr 2002 44:339-42. [Google Scholar]
[16]. Walter JH, Wraith JE, Cleary MA, Absence of acidosis in initial presentation of propionic acidemiaArch Dis Child 1995 72:197-99. [Google Scholar]
[17]. Abdelkreem E, Otsuka H, Sasai H, Aoyama Y, Hori T, Abd El Aal M, Beta-ketothiolase deficiency:Resolving challenges in diagnosisJ Inborn Errors Metab Screen 2016 4:01-09. [Google Scholar]
[18]. Mohammad SA, Abdelkhalek HS, Ahmed KA, Zaki OK, Glutaric aciduria type 1:Neuroimaging features with clinical correlationPediatr Radiol 2015 45:1696-705. [Google Scholar]
[19]. Nunes J, Loureiro S, Carvalho S, Pais RP, Alfaiate C, Faria A, Brain MRI findings as an important diagnostic clue in glutaric aciduria type 1Neuroradiol J 2013 26(2):155-61. [Google Scholar]
[20]. Simons A, Eyskens F, Glazemakers I, van West D, Can psychiatric childhood disorders be due to inborn errors of metabolism?Eur Child Adolesc Psychiatry 2017 26(2):143-54. [Google Scholar]
[21]. Ghaziuddin M, Al-Owain M, Autism spectrum disorders and inborn errors of metabolism:an updatePediatr Neurol 2013 49:232-36. [Google Scholar]
[22]. Zecavati N, Spence S, Neurometabolic disorders and dysfunction in autism spectrum disordersCurr Neurol Neurosci Rep 2009 9:129-36. [Google Scholar]
[23]. Manzi B, Loizzo AL, Giana G, Curatolo P. Autism and metabolic diseasesJ Child Neurol 2008 23:307-14. [Google Scholar]
[24]. Al-Owain M, Kaya N, Al-Shamrani H, Al-Bakheet A, Qari A, Al-Muaigl S, Autism spectrum disorder in a child with propionic academiaJIMD rep 2013 7:63-66. [Google Scholar]
[25]. Antshel K, ADHD, learning, and academic performance in phenylketonuriaMol Gen Met 2010 99:S52-S5. [Google Scholar]
[26]. Weber P, Scholl S, Baumgartner ER, Outcome in patients with profound biotinidase deficiency:Relevance of newborn screeningDev Med Child Neurol 2004 46:481-84. [Google Scholar]
[27]. Taitz LS, Green A, Strachan I, Bartlett K, Bennet M, Biotinidase deficiency and the eye and earLancet 1983 2:918 [Google Scholar]
[28]. Wolf B, Heard GS, Weissbecker KA, McVoy JR, Grier RE, Leshner RT, Biotinidase deficiency:initial clinical features and rapid diagnosisAnn Neurol 1985 18(5):614-17. [Google Scholar]
[29]. Wolf B, Spencer R, Gleason T, Hearing loss is a common feature of symptomatic children with profound biotinidase deficiencyJ Pediatr 2002 140(2):242-46. [Google Scholar]
[30]. Venkataraman V, Balaji P, Panigrahi D, Jamal R, Biotinidase deficiency in childhoodNeurol India 2013 61:411-13. [Google Scholar]