Paediatric Peripheral Primitive Neuroectodermal Tumour – A Clinico-Pathological Study from Southern India
Rithika Rajendran1, Leena Dennis Joseph2, Thanka Johnson3, Latha Magatha Sneha4, Julius Xavier Scott5, Satish Srinivasan6
1 Phd Scholar, Department of Pathology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
2 Professor, Department of Pathology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
3 Professor, Department of Pathology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
4 Assistant Professor, Division of Paediatric Haemato-Oncology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
5 Chief, Division of Paediatric Haemato-Oncology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
6 Associate Professor, Department of Radiation Oncology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, Chennai, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Leena Dennis Joseph, Professor, Department of Pathology, Sri Ramachandra Medical College and Research Institute, Sri Ramachandra University, No.1, Ramachandra Nagar, Porur, Chennai-600116, Tamil Nadu, India.
E-mail: leenadj@gmail.com
Introduction
Primitive Neuroectodermal Tumour (PNET)/Ewing Sarcomas (ES) are aggressive childhood malignancies with neuroectodermal differentiation.
Aim
To study the clinical presentation, morphology, Immun-ohistochemistry (IHC), management and outcome of all the cases of paediatric pPNET/ES reported in our tertiary care centre over a period of six years.
Materials and Methods
This was a retrospective study conducted at Sri Ramachandra Medical College and Research Institute, Chennai, India. All biopsy proven cases of peripheral PNET/ES, in patients less than 18 years of age for a period of six years were included in this study. The corresponding clinical details regarding initial presentation, treatment and follow up were retrieved from the case files and analysed. Survival rate was calculated and Kaplan-Meier survival curve was plotted.
Results
We describe eleven cases of paediatric peripheral PNET/ES. The mean age at presentation was 94.08 (±58.27) months with a male/female ratio of 1.2:1. About 27.3% cases, all male with a mean age of 140 months at presentation, had distant metastasis during initial diagnosis. Biopsy showed small round blue cell morphology on light microscopy. IHC revealed strong membranous staining for CD99 in all cases. All children were treated with neo-adjuvant chemotherapy and then surgery, followed by radiotherapy if indicated. The cases were followed up for a mean duration of 20.82 months (ranging from one to 66 months). Nine children are doing well on follow up (81.8% survival rate). Two cases with metastasis at initial presentation died. Patients with metastatic disease exhibited a mean duration of survival of 9.66 (±7.24) months and those with localized disease exhibited a mean duration of survival of 25 (±22.88) months.
Conclusion
Metastasis at diagnosis is the single most important factor affecting prognosis. This was reflected in the present study where cases with metastasis exhibited a short mean duration of survival when compared to localized disease. It is likely that many cases of PNET/ES were not accurately identified in the past as IHC plays a vital role in the diagnosis of these small round blue cell tumours. IHC in adjunct with molecular studies has improved diagnostic accuracy. Multidisciplinary management and good supportive care when the lesion is localized has lead to improved survival.
Children, CD99, Ewing sarcoma, Survival
Introduction
Peripheral PNET/ES falls under the category of PNET family of tumours. They are small round blue cell tumours with varying degrees of neuroectodermal differentiation occurring in the bone and soft tissues with common chromosomal translocations. Peripheral PNET (pPNET)/ES are quite rare and comprise 5%-10% of paediatric bone tumours and 4%-17% of paediatric soft tissue tumours [1,2]. They tend to have an aggressive clinical course with an overall three year survival rate of around 56%-65% [3]. In the past 10-15 years, the advent of IHC and advances in molecular studies has greatly enhanced the scientific knowledge regarding the tumour biology of PNET family of tumours. This has enabled improved diagnostic accuracy and treatment options with better survival outcomes.
Hence, the present study was done to evaluate the clinical presentation, morphology, IHC, management and outcome of all the cases of paediatric pPNET/ES reported in our tertiary care centre over a period of six years.
Materials and Methods
This was a record based retrospective study conducted at Sri Ramachandra Medical College and Research Institute, Chennai with Institutional Ethical Committee clearance. All biopsy proven cases of peripheral PNET/ES, in patients less than 18 years of age for a period of Jan 2010 to Dec 2015 were included in this study. A diagnosis of PNET/ES was made if the tumour cells showed diffuse, strong membranous staining for CD99. Positive controls were used. Cases with CD99 negative and those older than 18 years were excluded. The corresponding clinical details regarding initial presentation, treatment and follow up were retrieved from the case files and analysed. Survival rate was calculated and Kaplan-Meier survival curve was plotted. During the study period there were a total of 140 paediatric solid tumours of which 76 were small round blue cell tumours (54.3%). A total of 11 cases (14.6% of paediatric solid tumours) met the selection criteria and were included in our study.
Results
The clinical profile is shown in [Table/Fig-1]. The patients’ ages ranged from 3 months to 17 years. The mean age at presentation was 94.08 (±58.27) months with a male/female ratio of 1.2:1 (six boys and five girls). The girls presented earlier at a mean age of 53.4 months (ranging from 3 to 72 months) whereas the boys presented at a mean age of 128 months (ranging from 48 to 204 months). The majority of the tumours (63.6%) were located in the trunk (seven cases) followed by two each in humerus and femur. Eight children presented with localized disease. Three patients, all male, (27.3% of cases) presented with metastasis at the time of initial diagnosis.
Summary of clinical and histopathological details and follow up.
| S No | Gender/Age (years) | Primary site | Staging | Light microscopy | Immunohistochemistry (Positive) | Chemotherapy | Surgery | Radiation | Outcome | Duration of follow up (months) |
|---|
| 1 | M/12 | Left 2nd rib | Localized | SRBCT | Vm, CD99 | VDC & IE | Total excision of left 2nd rib | Nil | Alive and well | 36 |
| 2 | F/4 | Dorso lumbar region D10-L2 | Localized | SRBCT | Vm, CD99,S100 | VDC & IE | Excision biopsy | Nil | Alive and well | 48 |
| 3 | F/6 | Right humerus | Localized | SRBCT | Vm, CD99 | VDC & IE | Local resection | Nil | Alive and well | 66 |
| 4 | M/11 | Left femur | Metastatic(pulmonary metastasis) | SRBCT | Vm, CD99 | VIDE & Topotecan | Local resection | 50 Gys to primary site | Died due to relapse | 6 |
| 5 | M/13 | Left 4th rib | Localized | SRBCT | Vm, CD99 | VDC & IE | Excision of left 4th rib | 54 Gys to primary site | Alive and well | 1 |
| 6 | M/17 | Left hip joint | Metastatic(pulmonary metastasis) | SRBCT | CD99 | VIDE & Topotecan | Nil | 54 Gys to left pelvis and whole lung | Alive and well | 18 |
| 7 | F/6 | Left 5th rib | Localized | SRBCT | Vm, CD99 | VDC & IE | Excision of left 5th rib | 50 Gys to primary site | Alive and well | 15 |
| 8 | M/4 | Right 11th and 12th rib | Localized | SRBCT | Vm, CD99, Syn | VDC & IE | Excision of right 11th and 12th rib | 50 Gys to primary site | Alive and well | 18 |
| 9 | F/ 3 months | Left suprascapular region | Localized | SRBCT | Vm, CD99 | VDC & IE | Resection of mass | Nil | Alive and well | 12 |
| 10 | F/6 | Abdomen | Localized | SRBCT | Vm, CD99 | VDC & IE | Resection of mass | Nil | Alive and well | 4 |
| 11 | M/7 | Left humerus | Metastatic | SRBCT | Vm, CD99 | VIDE & Topotecan | Nil | Nil | Progressive disease, on palliative care, died | 5 |
SRBCT – Small round blue cell tumour; Vm – Vimentin; Syn – Synaptophysin
V - Vincristine, D - Doxorubicin, C - Cyclophosphamide, I – Ifosfamide, E – Etoposide
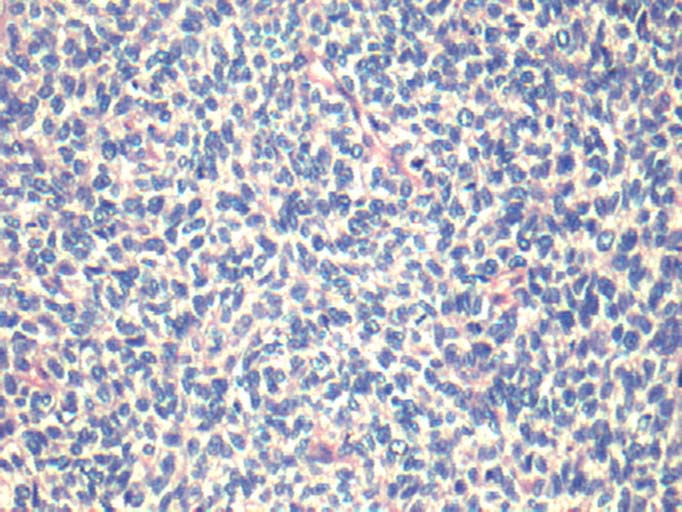
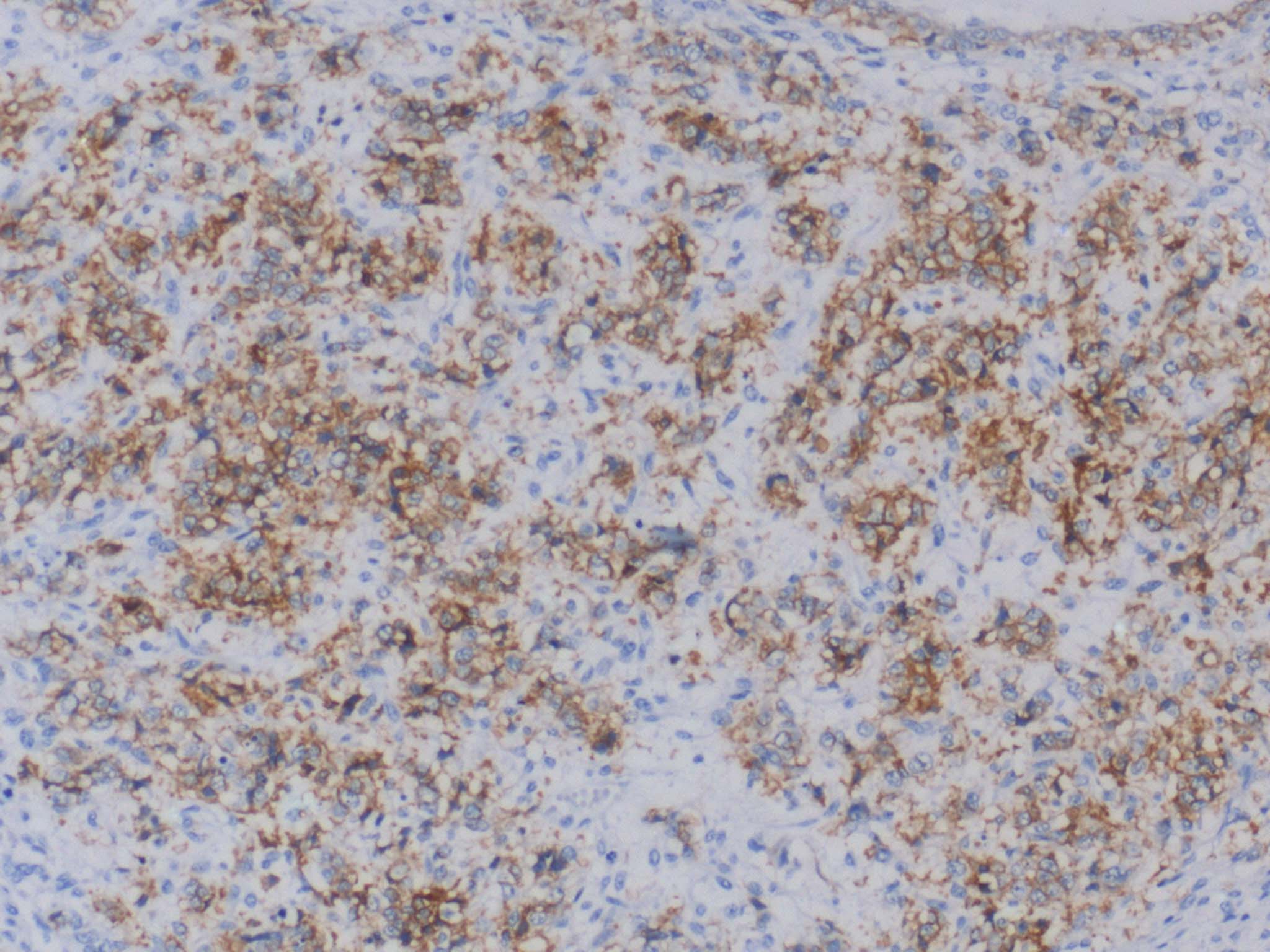
By light microscopy the tumours showed diffuse sheets and focal nests of monotonous small round blue cells with scant cytoplasm [Table/Fig-2]. There was increased and atypical mitosis and many apoptotic bodies. The common differential diagnoses on light microscopy included lymphoma, neuroblastoma, embryonal rhabdomyosarcoma, small cell osteosarcoma and pPNET/ES based on the location and age. For every case a choice of IHC markers was made from a panel which included vimentin, CD45, CD34, CD99, desmin, myogenin, S100, BCL2, synaptophysin, chromogranin and cytokeratin. All the cases were unequivocally positive for CD99 by IHC [Table/Fig-1,3].
Light microscopy shows sheets of monotonous small round blue cells (Haematoxylin and Eosin, 20X).
IHC shows diffuse membranous positivity for CD99 (10X).
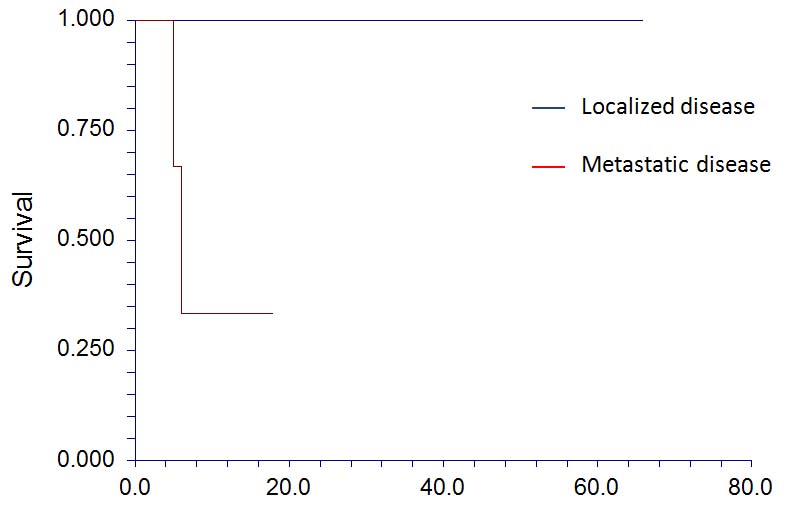
The children were managed with combination chemotherapy, surgery and radiation. All patients received 12 weeks of neo-adjuvant chemotherapy consisting of Vincristine, Doxorubicin and Cyclophosphamide (VDC) alternating with Ifosfamide and Etoposide (IE) or topetecan followed by resection of the tumour and adjuvant radiotherapy if required. Mean follow up duration was 20.82 months (minimum 1 month, maximum 66 months). Nine children are alive and well and are on regular follow up. Two children, who presented with metastatic disease, died at 5 months and 6 months after diagnosis due to progressive disease and relapse respectively (18.2% mortality). The Kaplan-Meier survival curve is shown in [Table/Fig-4].
Kaplan-Meier survival curve for localized and metastatic pPNET.
Discussion
The PNET family of tumours are small round cell tumours that have varying degrees of neuroectodermal differentiation. They can be classified into three groups namely Central Nervous System (CNS) PNET (arising in central nervous system), neuroblastoma (arising in autonomic nervous system) and peripheral PNET (pPNET) (arising from tissues outside the central and autonomic nervous system) [4]. Peripheral PNETs include Ewing sarcoma (osseous and extraosseous), malignant peripheral PNET or peripheral neuroepithelioma of bone and soft tissues and Askin tumour (PNET/ES of thoracopulmonary region) [5].
The PNET family of tumours is an uncommon and aggressive tumour of childhood and adolescence with an annual incidence of 2.93 per million populations [6]. The incidence rates have significantly increased over the last 25 years which may be attributed to improved diagnostic modalities [7]. Worldwide it is more common in Caucasians and less reported in the Asian population [8]. The incidence does not vary significantly according to sex but studies have reported a slight male preponderance which was also the case in our study [7,9].
Several studies have reported that pPNETs are most common in the thoracopulmonary region followed by the pelvis and extremities [3,10,11]. They are less common in the head and neck region. The site of the tumour may have a minor prognostic significance with patients having paraspinal and scapular disease faring the best, head and neck disease showing intermediate and abdominopelvic disease faring the worst [12]. In our study, the patient number was too small to conclude the effect of tumour site on survival. Both patients who died had metastatic disease at presentation hence, the effect of local site could not be determined.
It is likely that the diagnosis of PNET/ES was under-reported till the advent of IHC which enabled the diagnosis of poorly differentiated small round cell tumours. IHC profile is vital to distinguish pPNETs from other small round blue cell tumours as it affects management and prognosis. Peripheral PNETs typically express membrane positivity for CD99 which is the gene product for MIC2 [12]. It is highly sensitive but not specific for pPNET as it is frequently expressed in other small round blue cell tumours as well. However, the pattern of staining is often cytoplasmic rather than the typical membranous staining observed here [13]. pPNETs frequently co-express vimentin as well. Other nonspecific markers reported include S100, synaptophysin, neuron specific enolase and CD75 [14]. All eleven cases studied showed strong membranous CD99 and ten showed vimentin immunopositivity. One case each showed focal positivity for S100 and synaptophysin respectively. CNS PNETs lack CD99 expression [4,15,16].
Molecular studies show that all these tumours share the common reciprocal translocation of chromosomes 11 and 22. All PNET family of tumours express Friend Leukaemia virus Integration-1 (FLI-1)which is the fusion product of Ewing sarcoma gene (22q12) and FLI-1 (11q24). It is very sensitive and specific and can be detected by IHC as well as molecular methods [17,18]. Cytogenetic analysis reveals that this reciprocal translocation t(11;22) is present in approximately 85% of the cases while the remaining 5%-10% show translocation between EWS and ERG (ETS related gene) (21q22) [19,20]. Cytogenetic analysis was not done in our study taking into account the socioeconomic background of the majority of the patients.
Several studies have shown that 26%-28% of children present with distant metastasis at initial presentation [8]. This was observed in 27.3% cases in our study population as well. Reverse Transcription PCR (RT-PCR) analysis has shown that up to 30% of patients with clinically localized disease have micrometastasis in the bone marrow [21].
Current treatment recommendations advocate multimodal approach including surgical resection, adjuvant and neoadjuvant chemotherapy and radiotherapy for both localized and metastatic disease [22]. The most recommended chemotherapeutic regimen includes vincristine, doxorubicin and cyclophosphamide with ifosfamide and etoposide [23]. Complete surgical resection is vital for better survival outcomes. Long term follow up of pPNET/ES survivors treated with radiotherapy shows a six fold risk for subsequent cancers when compared to the general population [24]. This includes secondary sarcomas, acute myeloid leukaemia and myeloblastic syndromes. This appears to be radiation dose dependant and it is shown that those exposed to less than 48 Gy had no additional risk [25]. High doses of etoposide have been implicated in the incidence of secondary leukaemia reported in 1%-2% of treated cases [26]. None of our patients presented with treatment related secondary malignancies during the study period. Since it has a low incidence, a bigger study population as well as a long duration of follow up is required to comment on the same.
The most significant prognostic factor is the presence of distant metastasis [27]. In the present study, 100% cases (8/8) with localized disease were alive and well on follow up, with a mean duration of survival of 25 (± 22.88) months. Cases with metastatic disease exhibited a poor outcome with 66.7% cases (2/3) died and a short duration of survival (mean 9.66 (±7.24) months). The primary site did not have any significant effect on the outcome in our study. The Kaplan-Meier survival curve is shown in [Table/Fig-4].
Limitation
The study however is limited by the small number of patients as well as the short duration of follow up (mean follow up duration of 20.82 months, ranging from minimum one month to maximum 66 months).
Conclusion
An early accurate diagnosis of peripheral PNET is very essential as the presence of metastasis is the most important factor adversely affecting survival. This was reflected in the present study where patients with metastasis exhibited a poor outcome with (two out of three cases dead) and a short duration of survival (mean 9.66 (±7.24) months) when compared to localized disease with 100% cases alive and well on follow up. Improved diagnostic modalities including immunohistochemistry and molecular techniques have played a vital role in early diagnosis. Multidisciplinary management and good supportive care when the lesion is localized has lead to improved survival.
SRBCT – Small round blue cell tumour; Vm – Vimentin; Syn – Synaptophysin
V - Vincristine, D - Doxorubicin, C - Cyclophosphamide, I – Ifosfamide, E – Etoposide
[1]. Siegel DA, King J, Tai E, Buchanan N, Ajani UA, Li J, Cancer Incidence Rates and Trends Among Children and Adolescents in the United States, 2001-2009Paediatrics 2014 134:e945-55. [Google Scholar]
[2]. Windfuhr J, Primitive neuroectodermal tumour of the head and neck: incidence, diagnosis, and managementAnn Oto Rhino Laryngo 2004 113:533-43. [Google Scholar]
[3]. Tsokos M, Alaggio R, Dehner L, Dickman P, Ewing sarcoma/peripheral primitive neuroectodermal tumour and related tumoursPed Dev Pathol 2012 15:108-26. [Google Scholar]
[4]. Batsakis J, MacKay B, El-Naggar A, Ewing’s sarcoma and peripheral primitive neuroectodermal tumour: an interim reportAnn Oto Rhino Laryngo 1996 105:838-43. [Google Scholar]
[5]. Fletcher CDM, Unni KK, Mertens F, World Health Organisation Classification of tumours: Tumours of Soft Tissue and Bone 2002 GenevaWHO Press:298 [Google Scholar]
[6]. Esiashvili N, Goodman M, Marcus R, Changes in Incidence and Survival of Ewing Sarcoma Patients Over the Past 3 DecadesJ Ped Hemat Onco 2008 30:425-30. [Google Scholar]
[7]. Toro J, Travis L, Wu H, Zhu K, Fletcher C, Devesa S, Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26,758 casesInt J Cancer 2006 119:2922-30. [Google Scholar]
[8]. Stiller C, McKinney P, Bunch K, Bailey C, Lewis I, Childhood cancer and ethnic group in Britain: a United Kingdom children’s Cancer Study Group (UKCCSG) studyBr J Cancer 1991 64:543-48. [Google Scholar]
[9]. Dennis NF, Francis M, Lawrence G, Soft tissue sarcoma incidence and survival: tumours diagnosed in England between 1985 and 2009United Kingdom: National Cancer Intelligence Network 2012 :38-42. [Google Scholar]
[10]. Kushner B, Hajdu S, Gulati S, Erlandson R, Exelby P, Lieberman P, Extracranial primitive neuroectodermal tumours. The memorial sloan-kettering cancer center experienceCancer 1991 67:1825-29. [Google Scholar]
[11]. Marina NM, Etcubanas E, Parham DM, Bowman LC, Green A, Peripheral primitive neuroectodermal tumour (peripheral neuroepithelioma) in children. A review of the St. Jude experience and controversies in diagnosis and managementCancer 1989 64:1952-60. [Google Scholar]
[12]. Halliday BE, Slagel DD, Elsheikh TE, Silverman JF, Diagnostic utility of MIC-2 immunocytochemical staining in the differential diagnosis of small blue cell tumoursDiag Cytopathol 1998 19:410-16. [Google Scholar]
[13]. Ginsberg JP, de Alava E, Ladanyi M, Wexler LH, Kovar H, Paulussen M, EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcomaJ Clin Oncol 1999 17:1809 [Google Scholar]
[14]. Folpe AL, Hill CE, Parham DM, O’Shea PA, Weiss SW, Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumours with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumourAmerican J Surg Pathol 2000 24:1657-62. [Google Scholar]
[15]. Dedeurwaerdere F, Giannini C, Sciot R, Rubin BP, Perilongo G, Borghi L, Primary peripheral PNET/Ewing’s sarcoma of the dura: a clinicopathologic entity distinct from central PNETModern Pathol 2002 15:673-78. [Google Scholar]
[16]. Le Deley MC, Delattre O, Schaefer KL, Burchill SA, Koehler G, Hogendoorn PC, Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumour : prospective results from the cooperative Euro-EWING 99 trialJ Clin Oncol 2010 28:1982-88. [Google Scholar]
[17]. Ishii N, Hiraga H, Sawamura Y, Shinohe Y, Nagashima K, Alternative EWS-FLI1 fusion gene and MIC2 expression in peripheral and central primitive neuroectodermal tumoursNeuropathology 2001 21:40-44. [Google Scholar]
[18]. Bao-yue LI, Yu YA, Juan DU, Yan ZH, Hua WA, Jie ZH, Application of the in situ hybridization with EWS dual-color break-apart fluorescence probe and anti-CD99 and anti-FLI-1 antibodies in the diagnosis of Ewing’s sarcoma/primitive neuroectodermal tumourJ Peking University (Health Sciences) 2008 4:006 [Google Scholar]
[19]. Ladanyi M, EWS-FLI1 and Ewing’s sarcoma: recent molecular data and new insightsCancer Bio Therapy 2002 1:329-35. [Google Scholar]
[20]. May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, Ewing sarcoma 11; 22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformationProceedings of the National Academy of Sciences 1993 90:5752-56. [Google Scholar]
[21]. Athale UH, Shurtleff SA, Jenkins JJ, Poquette CA, Tan M, Downing JR, Use of reverse transcriptase polymerase chain reaction for diagnosis and staging of alveolar rhabdomyosarcoma, Ewing sarcoma family of tumours, and desmoplastic small round cell tumourJ Ped Hematol Oncol 2001 23:99-104. [Google Scholar]
[22]. Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, Ewing’s sarcoma family of tumours: current managementOncologist 2006 11:503-19. [Google Scholar]
[23]. Carvajal R, Meyers P, Ewing’s sarcoma and primitive neuroectodermal family of tumoursHematology/oncology clinics of North America 2005 19:501-25. [Google Scholar]
[24]. Inskip PD, Curtis RE, New malignancies following childhood cancer in the United States, 1973-2002Int J Cancer 2007 121:2233-40. [Google Scholar]
[25]. Saenz NC, Hass DJ, Meyers P, Wollner N, Gollamudi S, Bains M, Paediatric chest wall Ewing’s sarcomaJ Ped Surg 2000 35:550-55. [Google Scholar]
[26]. Paulussen M, Ahrens S, Lehnert M, Taeger D, Hense HW, Wagner A, Second malignancies after Ewing tumour treatment in 690 patients from a cooperative German/Austrian/Dutch studyAnn Oncol 2001 12:1619-30. [Google Scholar]
[27]. Paulussen M, Ahrens S, Burdach S, Craft A, Dockhorn-Dworniczak B, Dunst J, Primary metastatic (stage IV) Ewing tumour: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma StudiesAnn Oncol 1998 9:275-81. [Google Scholar]